
Cell重磅揭示前列腺癌中circRNA新的调控机制
信息来源:金开瑞 作者:genecreate 发布时间:2019-04-18 15:57:09
题目:Widespread and functional RNA circularization in localized prostate cancer
期刊:Cell
影响因子:31.2
主要技术:circRNA-Seq / circRNA pull-down
研究背景
前列腺癌是全世界男性最常见的恶性肿瘤之一,一旦肿瘤迁移到腺体外就不可避免地会出现无法治愈的转移性疾病。因此,在临床上研究局部可治愈的前列腺肿瘤的生物学特性是非常有必要的。
circRNA是一个稳定的共价闭合环,可躲避poly-A的捕获,具有创造生物标志物的潜力。虽然已经有研究者挖掘癌症基因组图谱网络和评估组学信息,但是前列腺癌转录组的完全多样性及其意义仍然是未知的。
本文作者选取了临床144例局限性前列腺肿瘤样本进行超深度RNA测序,共识别了1233个独特的融合基因和76311个不同的circRNAs,并确定了171个对前列腺癌细胞增殖至关重要的circRNAs。由此可见,本次转录组数据中包含许多传统分子策略无法观察到的功能实体,为将来RNA转录组研究提供了丰富的资源。

研究内容及结果
1. 局限性前列腺癌的转录组分析
本文对144例前列腺肿瘤样本进行转录组分析,发现在所有肿瘤中有18340个转录本(图1A-1B,其中包含15112个蛋白质编码亚型)。结合癌症基因组图谱网络,按四分位间距排列前25% RNA并进行聚类分析,发现4585个转录本中包含3905个编码RNA和680个非编码RNA(图1C),并描述了5个患者亚型(P1-P5)和5个RNA亚型(R1-R5)。
为确定候选肿瘤特异性亚型,从而评估融合基因的结构以及与侵袭性局部疾病的关系,发现有21例复发率超出预期的复发性融合(图1D),并在超过10% SU2C转移转录本中发现复发融合(图1E,有17个高丰度的融合物,有12个在转移病灶中复发)。结果揭示融合基因的转变可能预示着对治疗效果、转移部位和疾病进展的选择性适应。
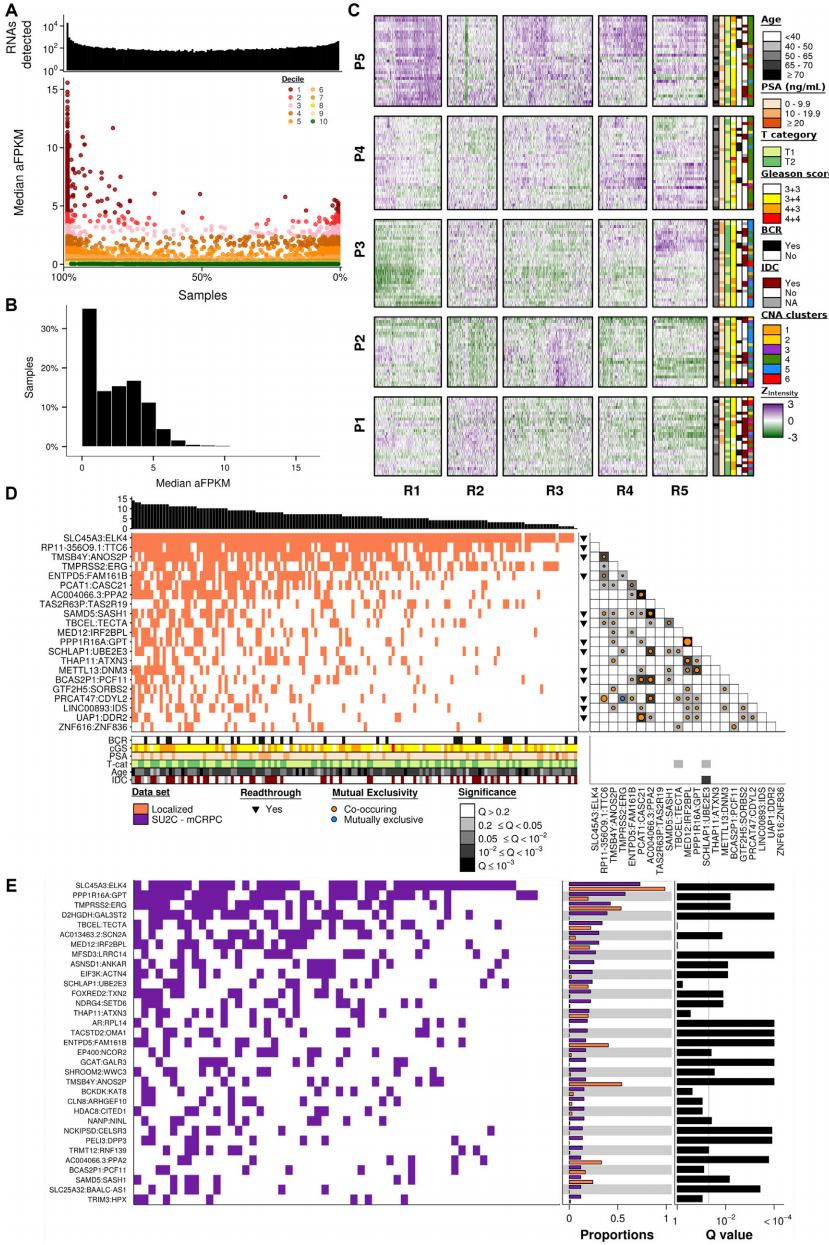
图1 局限性前列腺癌的转录组学特征
2. 局限性前列腺癌的circRNA表达谱
作者针对前列腺癌患者的肿瘤和细胞系展开序列分析和联合治疗,确定了34286个circRNAs,其中有33806个在RNase R处理后至少增加了1.5倍(图2A)。将重叠区域中高信度的25036个circRNAs与circBase注释进行比较,发现11例细胞或组织源中仅有1例检测到67%的circRNAs(图2B-2C)。
在基因水平上检测每个样本的亲本线性、全线性和环状的分子分数,其亲本线性形式的高值表明了几乎所有肿瘤的基因都有产生circRNA的倾向(图2D)。采用STAR方法评估每个基因的八个特征并量化其环化的能力,结合ROC曲线来预测RNA环化事件并分析与丰度的相关性,结果表明circRNA环化产生所需的一些信息被嵌入到基因组中。高丰度circRNA往往具有高丰度线性亲本RNA对应物(图2E-2F),统计发现有127个circRNA的丰度显著地高于线性对应结构(图2H)。
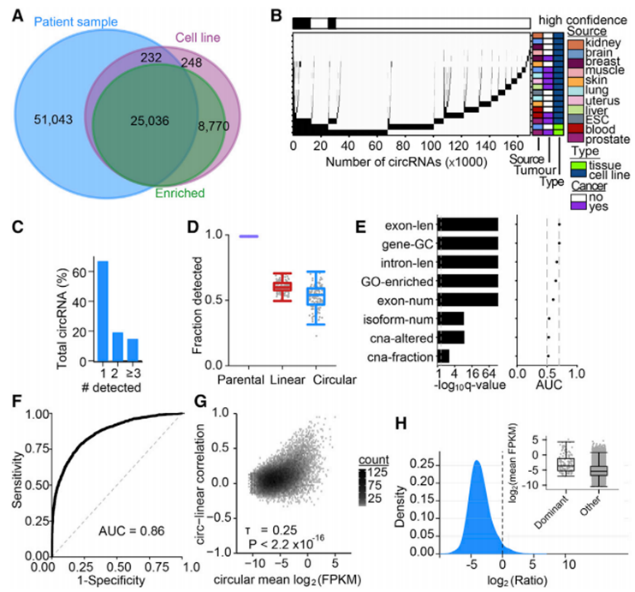
图2 前列腺癌中circRNAs的鉴定与表征
3. circRNA丰度定义临床上不同的肿瘤亚型
将144例肿瘤患者样本分成四组份,采用circRNA-Z评分来量化特定的肿瘤亚型(图3A)。通过比较CPC-GENE和NGS-ProToCol队列发现,在预后水平上极端(Q1/Q4)患者组的circRNA分布情况明显弱于稳定(Q2/Q3)患者组(图3B- 3C),且伴随着较高的转移发生率(图3D)。通过比对CPC基因数据集中转录丰度与BCR注释关系(图3E),发现总circRNA和特异circRNA丰度均与临床结果相关,重点提示了circRNA并非是转录噪音而是可能具有特定的致癌功能。
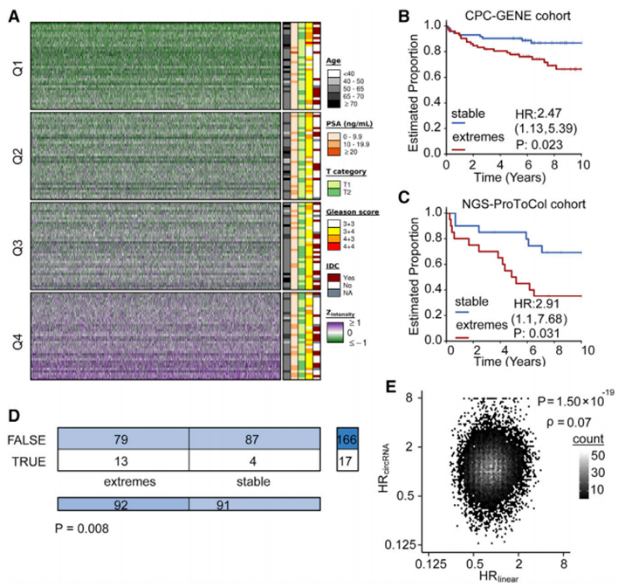
图3 circRNA失调与前列腺癌进展有关
4. 功能筛选识别关键的circRNA
如图4A中shRNA定位策略示意图所示,通过后剪切位点设计shRNAs来筛查circRNA的特异性功能,由此针对1507个circRNA和1075个线性转录物均至少配有2个shRNAs(图4B)。采用MaGeCK来鉴定耗尽的线性转录物和提高筛选有效性,发现在V16A细胞系中阳性对照组明显高于阴性对照组,归一化后shRNA丰度变化与时间点呈现高度相关(图4C-4D)。由此逐步确定,在一个细胞系中总共至少有171个关键的circRNAs,其中91.8% circRNAs是必需的而其线性对应物是非必需的(图4E-4F)。同时在四个细胞系中对10个关键的circRNAs进行敲除(KD)和过表达(OE)后测定细胞增殖情况,并结合LNCaP CRISPR数据库筛查(图4G-4H),发现circRNA驱动细胞增殖的过程独立于其线性对应物。
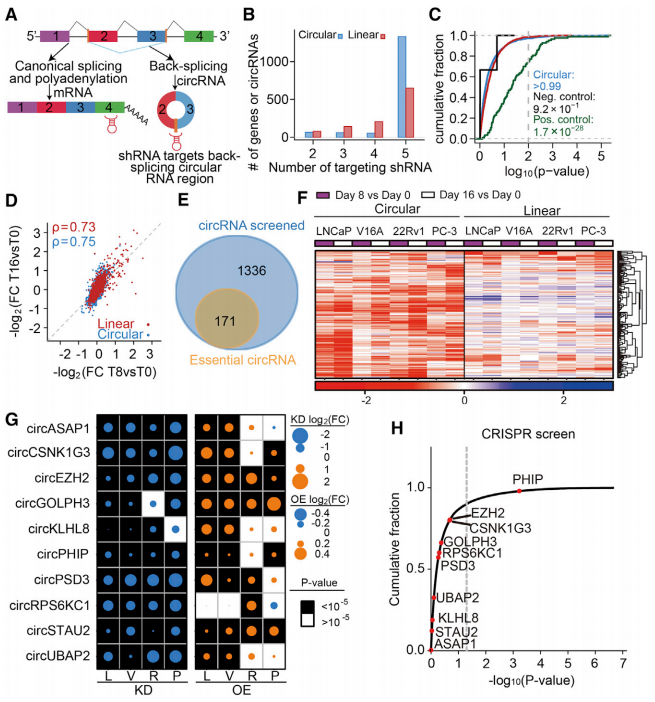
图4 shRNA筛选和关键circRNAs的验证
5. 关键circRNAs的功能表征
在10个关键的circRNAs中,circCSNK1G3总长度为536 bp且包含了三个外显子,在CPC基因队列中发现其与线性对应物的丰度相似(环状:线性 = 0.79,图5A和5B)。通过对circCSNK1G3-OE/-KD处理的PC-3细胞进行RNA序列分析,发现许多基因在OE样本和KD样本呈现反向变化趋势,并统计出279个激活基因和196个抑制基因(图5C),同时观察到在circCSNK1G3- KD样本中有环状和线性亚型共享的6个靶基因(图5D)。在测序报告注释后发现circCSNK1G3激活基因与细胞周期相关且促进细胞增殖,而CSNK1G3激活的关键是对酸性化学物质的反应(图5E-5F),这些数据均表明circCSNK1G3可促进前列腺癌细胞的增殖但不依赖于其线性转录本。
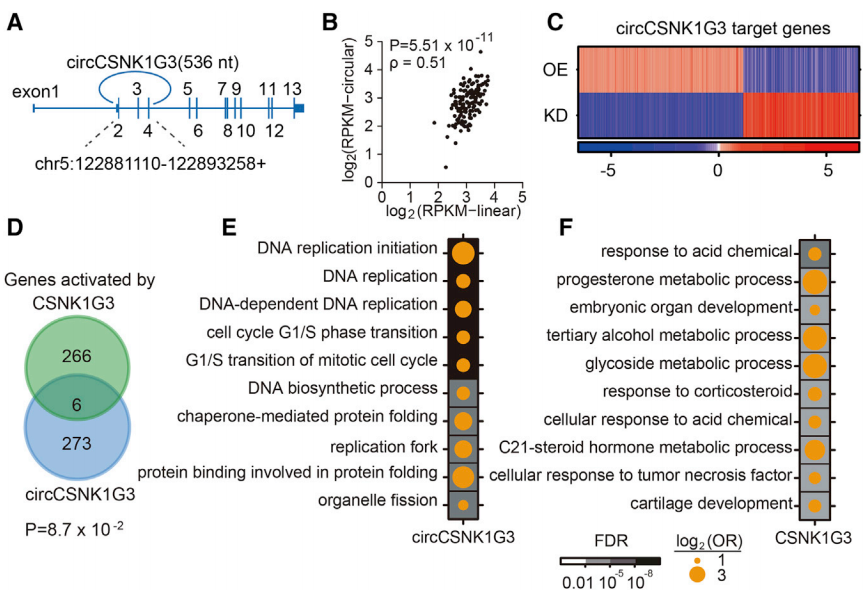
图5 circCSNK1G3的基础功能表征
6. circCSNK1G3与miR-181b/d互作促进前列腺癌细胞增殖
结合银酸交联和免疫沉淀测序(AGO-CLIP-Seq)数据进行分析预测,作者筛选出与circCSNK1G3互作的10个候选miRNAs。通过对CPC基因队列亚群(n=119)中miRNA数据集分析,发现有5个miRNA(miR-181a/b/c/d 和 miR-196b)与circCSNK1G3表达显著相关(图6A),并预测出高度饱和的AGO结合位点在外显子2区域(图6B)。接着通过生物素标记circCSNK1G3进行RNA pull-down实验,观察到只有miR-181b/d显著地富集(图6C),且miR-181b/d反向验证实验也证实了这一点(图6D)。在miR-181b/d-OE的PC-3细胞中发现细胞增殖和CBX7丰度显著地下调,在circCSNK1G3-KD验证实验中发现mir-181b/d显著地下调和cbx7显著地上调,推测出CBX7作为前列腺癌中miR-181b特征靶点和肿瘤抑制因子(图6E-6F)。与此同时,发现细胞周期基因(如CDK1和CDC25A)在miR-181b/d-OE细胞中上调且在circCSNK1G3-KD细胞中下调。综上所述,circCSNK1G3至少部分地参与miR-181b/d相互作用来促进前列腺癌细胞增殖。
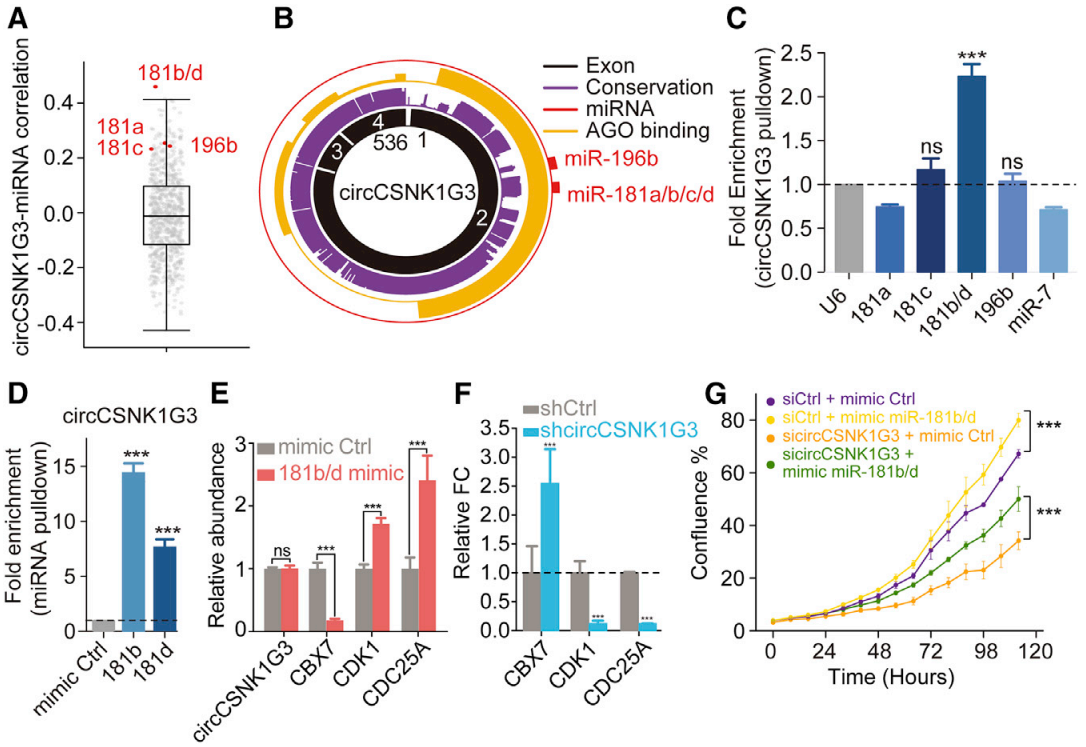
图6 miR-181b/d部分挽救了circCSNK1G3的丢失
文章小结
本文旨在探讨深层RNA序列中原发肿瘤的价值,并首次报道了具有功能注释的circRNAs,揭示了一种与侵袭性导管内癌组织学相关的线性转录亚型和一种区分局限性转移性疾病的融合特征,并为后期RNA转录组研究提供丰富的资源。
与此同时,反式剪切事件表明了广泛的RNA发生环化,在肿瘤细胞中平均有7232个circRNA表达,结合多个患者样本数据分析得到circRNA产生的程度与疾病进展相关。功能缺失筛查发现11.3% 的高丰度circRNA对细胞增殖至关重要,其中90%与线性转录本并不相关。不同circRNA具有不同的特异性功能,例如circCSNK1G3通过与miR-181相互作用促进肿瘤细胞增殖。综上所述,相对于线性转录本而言,circRNA显示出独特的本质信息,对后期临床研究具有重要的作用。
解析文献
Chen S, Huang V, Xu X, et al. Widespread and functional RNA circularization in localized prostate cancer. Cell. 2019 Feb 7; 176(4): 831-843.
最新动态
-
04.22
一文读懂EMSA技术核心要点,让“emsa” 秒变“easy”
-
04.02
4·2世界孤独症日 | 聆听“星”声“泌”语,让爱来,让碍走
-
04.01
酵母杂交核心技术:深度Q&A帮你轻松突破实验瓶颈
-
03.19
【客户文献解读,IF>11】食管癌的"隐形推手":MALR-ILF3-HIF1a轴的强大作用
-
03.18
siRNA介绍及药物研发的现状前景
-
03.18
知无不“研”|5篇高分文献带你一览高通量酵母杂交的非凡魅力~
-
02.27
【客户文章分享】SHMT2 通过 5′UTR 依赖性 ADAM10 翻译启动介导小分子诱导的阿尔茨海默病病理学缓解过程
-
01.24
客户文献解读 | 中医为什么能治流感?是玄学还是运气?INT J NANOMED揭示鱼腥草抗病毒机制及范围!
-
01.24
客户文献分享,IF>11|Shank3:脑缺血再灌注损伤的守护者,揭示神经保护的新篇章
-
12.27
文献解读 | 高密度脂蛋白通过miR-181a-5p调控自噬影响血管新生
 X
X 






