
表观遗传 | 超级增强子相关lncRNA如何影响肝癌
前言:增强子(enhancer),是距离启动子较远距离的一段可与蛋白质(反式作用因子,trans-acting factor)结合的区域,可以被转录因子等蛋白结合从而激活基因转录。增强子在不同类型细胞以及不同发育阶段会有不同活性,并且与转录因子(Transcript Factor, TF)等蛋白的结合也具有特异性,因此增强子是调控基因特异性表达重要的顺式作用元件(Cis-regulatory element)(Fig 1)[1]。
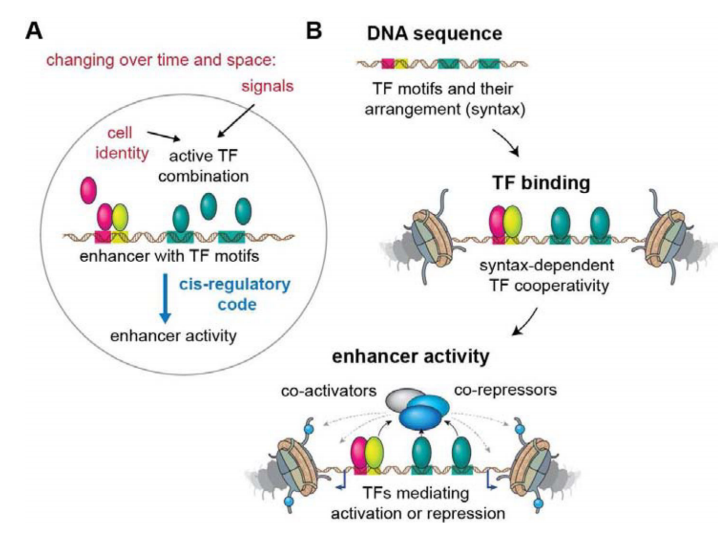
Figure 1 转录因子调控增强子活性示意图[1]。(A)在不同时空中,特定TF通过转录或细胞外信号进行识别motif序列与增强子结合并激活增强子。(B)顺式调控元件含有特定的TF基序(motif)。TF招募共激活因子或共阻遏因子,其调控增强子的活性。
在后生动物中,启动子具有H3K4me3标记,同样增强子在基因组上也有特殊的身份标记——增强子特征是具有染色质开放性和组蛋白H3K4me1修饰,活性增强子(Active Enhancer)具有H3K27ac标记,静态/起始增强子(Poised/Primed Enhancer)缺少H3K27ac标记(Fig 2)[2]。因此我们可以根据CUT&TAG、ChIP-seq和ATAC-seq等技术鉴定增强子位点。

Figure 2 顺式调控元件基因组特征[2]。增强子特征是具有染色质开放性和组蛋白H3K4me1修饰,活性增强子(Active Enhancer)具有H3K27ac标记,静态/起始增强子(Poised/Primed Enhancer)缺少H3K27ac标记。
随着对增强子在转录调控作用研究的深入,Richard A. Young团队于2013年提出超级增强子概念——超级增强子(Super-enhancer, SE)比普通增强子(Typical enhancer,TE)具有更高的转录激活相关组蛋白修饰(H3K27ac、H3Kme1等)、Mediator复合体和BRD4蛋白(Bromodomain containing 4)及转录因子的富集密度,超级增强子区域长度通常可达8-20 Kb,远高于普通增强子的200-300 bp长度[3]。长非编码RNA(lncRNA)的异常表达已在多种癌症中屡见报道,而SE与lncRNA关联的新研究思路近年来初露锋芒。SE-lncRNA就是超级增强子关联的lncRNA,它紧靠SE并且其转录活性受到SE的调控。这里小编分享2篇近期的肝癌方面SE-lncRNA高水平研究,帮助各位老师了解SE-lncRNA课题研究思路。
一、超级增强子驱动的lncRNA-Daw通过激活Wnt/b-catenin途径促进肝癌细胞增殖
广州中医药大学张锦芳团队2021年在Mol Ther Nucleic Acids. 发表Super-enhancer-driven lncRNA-DAW promotes liver cancer cell proliferation through activation of Wnt/β-catenin pathway一文揭示了lncRNA-DAW在肝癌发展中的作用机制[4]。
1、首先比较各人体组织中H3K27ac ChIP-seq数据,以筛选肝部特异性SE(Fig 3)。与其他实体瘤相似,在肝细胞癌(HCC)中也经常检测到基因组DNA拷贝数的扩增,包括20q13.12的扩增,既往研究表明,20q13.12片段的获得与肝癌患者的肝转移、总生存期及不良预后显著相关。而20q13.12片段在HCC中有比正常细胞更高的H3K4me1和H3K27ac信号,该位点可能含有肝脏特异性超级增强子。

Figure 3 人各组织H3K27ac数据[4]。(A) 一组正常人体组织中lncRNA DAW(LINC01430)基因座的H3K27ac-ChIP-seq数据。黄色区域表示lncRNA DAW的位点。(B) 肝细胞中lncRNA DAW基因座的超级增强子组蛋白标记的ChIP-seq数据。
2、在该SE区域鉴定到8个此前未报道的lncRNA,根据RNA-seq数据,此前被称为lncRNA-DAW的lncRNA在肝组织中高表达。比较肝细胞癌(HCC)和正常细胞中表达量,发现lncRNA-DAW显著上调。因此,作者将它确定后HCC候选靶点lncRNA进行进一步细胞和动物实验。
3、超级增强子经常被各种转录因子占据以发挥作用,作者筛选lncRNA-DAW所在SE依据UCSC基因组浏览器和Cistrome数据库候选结合的TF,发现该基因位点与肝细胞核因子家族成员HNF4G结合。根据ChIP-seq实验、双荧光素酶实验和RT-PCR实验,HNF4G 与候选区域中的SE-1 和 SE2结合,并且HNF4G促进lncRNA-DAW表达。
4、作者通过体外过表达和裸鼠实验证明lncRNA-DAW促进癌细胞增殖和转移。对lncRNA-DAW过表达细胞进行RNA-seq,对癌症相关通路中选择候选基因进行RT-qPCR实验和siRNA干扰实验,Wnt2基因表现受到lncRNA-DAW调控。
5、根据此前报道lncRNA可能与多梳蛋白抑制复合物(Polycomb repressive complexes, PRC)结合,作者发现PRC核心成分EZH2会抑制Wnt2表达。根据ChIP-seq实验发现,EZH2会结合Wnt2启动子从而抑制转录。
6、作者通过RIP-qPCR验证EZH2会结合lncRNA-DAW,lncRNA-DAW过表达后EZH2对Wnt2启动子抑制效果下降。由于过去报道多种lncRNA可以调节RNA结合蛋白的磷酸化和稳定性,于是作者通过WB验证了lncRNA-DAW的瞬时过表达以剂量依赖性方式降低内源性EZH2蛋白量。
7、依据细胞和动物实验作者证明Wnt2激活了Wnt/β-catenin通路促进癌细胞增殖。作者根据50对肝癌表达谱数据发现HCC标本中lncRNA-DAW和Wnt2的表达之间的显著正相关,lncRNA-DAW和Wnt2表达的增加可能是人类HCC组织中的常见事件,表明lncRNA-DAW和Wnt2可能作为肝癌患者的假定治疗靶点(Fig 4)。
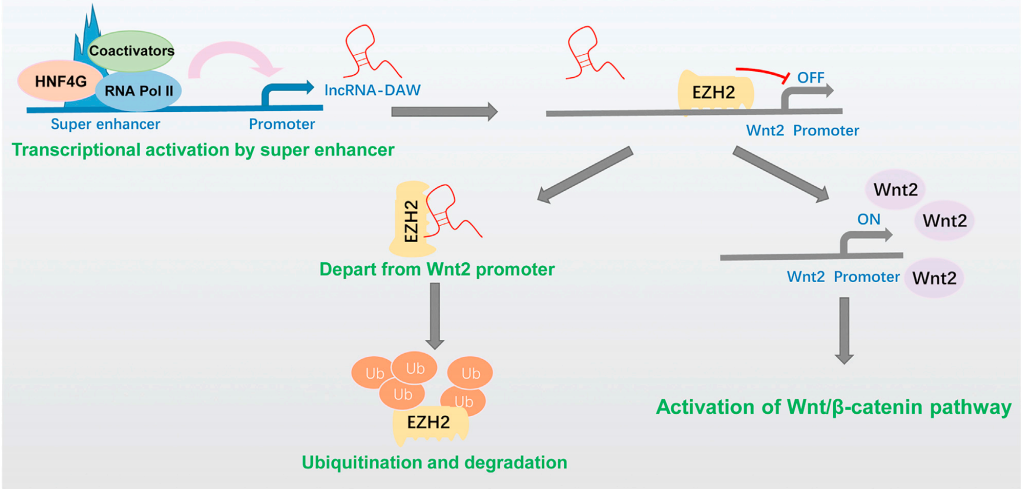
Figure 4 lncRNA-DAW调控Wnt2促进肝癌细胞增殖示意图[4]。
二、lncRNA FASRL促进脂肪酸生物合成并加剧肝癌进展
中山大学彭丽团队于去年在Advanced Science上发表了题为“Upregulation of Superenhancer-Driven LncRNA FASRL by USF1 Promotes De Novo Fatty Acid Biosynthesis to Exacerbate Hepatocellular Carcinoma”文章。研究发现了一种名为FASRL 的lncRNA表达受超级增强子调控,表达上调后可以促进脂肪酸的从头合成,从而促进肝细胞癌的发生[5]。
1、作者通过HepG2、Huh7、PLC、LM3(四种肝癌细胞系)和正常肝组织的H3K27ac数据,筛选并鉴定了由HCC细胞中异常超级增强子调节的lncRNA(Fig 5)。作者鉴定到31个肝癌特异性SE相关的lncRNA,其中3个lncRNA在HCC样本中上调,其高表达与HCC患者预后较差相关。作者将其中表达差异最大的1个lncRNA命名为 “FASRL”并作为候选肝癌靶点SE-lncRNA。FASRL位于16号染色体上,长度为15551 bp,包含两个外显子和一个内含子。
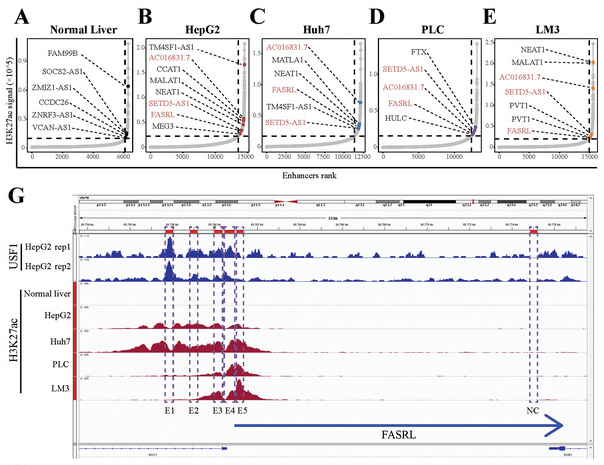
Figure 5 筛选肝癌特异性超级增强子[5]。A-E. 正常肝组织和四种肝癌细胞系超级增强子,FASRL是四种癌细胞系共有的特异性超级增强子,虚线代表ROSE软件判断SE的阈值。G. FASRL处H3K27ac信号,虚线为增强子位点,四种肝癌细胞中存在较高H3K27ac信号,而正常肝细胞中不存在。
2、作者将FASRL的五个增强子序列进行双荧光素酶实验,增强子E4对FASRL表达增强效果最强。为了探究SE调控FASRL表达上游机制,作者对FASRL序列进行转录因子motif分析,把将排名前十的TF表达量与FASRL表达量进行相关性分析,相关性最高的是热休克转录因子1(HSF1)。之后通过siRNA和ChIP-qPCR实验证明TF USF1可以通过与HCC中的SE结合来转录驱动FASRL表达(Fig 6)。
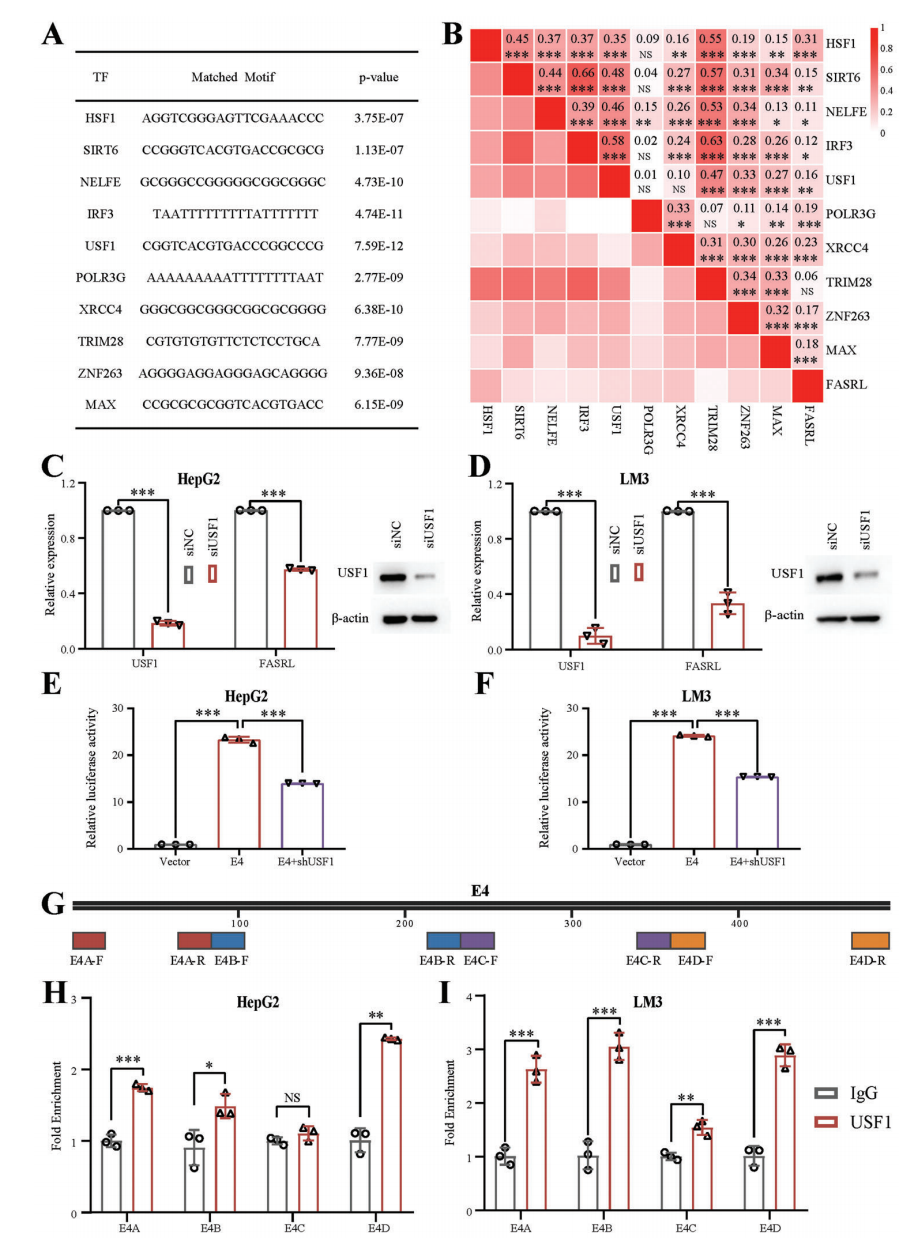
Figure 6 验证HSF1是调控FASRL的转录因子[5]。A. FASRL区域序列前10可能结合的TF motif序列。B. 10个TF和FASRL表达相关性。C,D. siRNA干扰USF1后HepG2和LM3细胞系中USF1和FASRL表达量和蛋白含量。E,F. 双荧光素酶实验显示了shRNA敲低USF1后E4的转录活性下降。G. 在E4序列上设计了4对ChIP-qPCR引物,H图为USF1结合SE的ChIP-qPCR结果。
3、作者进行体外实验验证FASRL是否能够影响肝癌细胞进展。通过siRNA敲低FASRL后细胞增殖、细胞迁移、细胞凋亡证明FASRL可以促进HCC细胞的致癌生长。之后将稳定转染shFASRL或shNC的HCC细胞系皮下注射到裸鼠中,观测到shFASRL显著减缓了裸鼠中肿瘤细胞的增殖。
4、为了揭示FASRL的特定下游调控途径,作者依据siFASRL转录组数据进行基因集富集分析(GSEA),FASRL敲低与脂肪酸(FA)途径相关基因的表达呈显著负相关,之后进行RT-qPCR和WB实验验证了这个发现(Fig 7)。
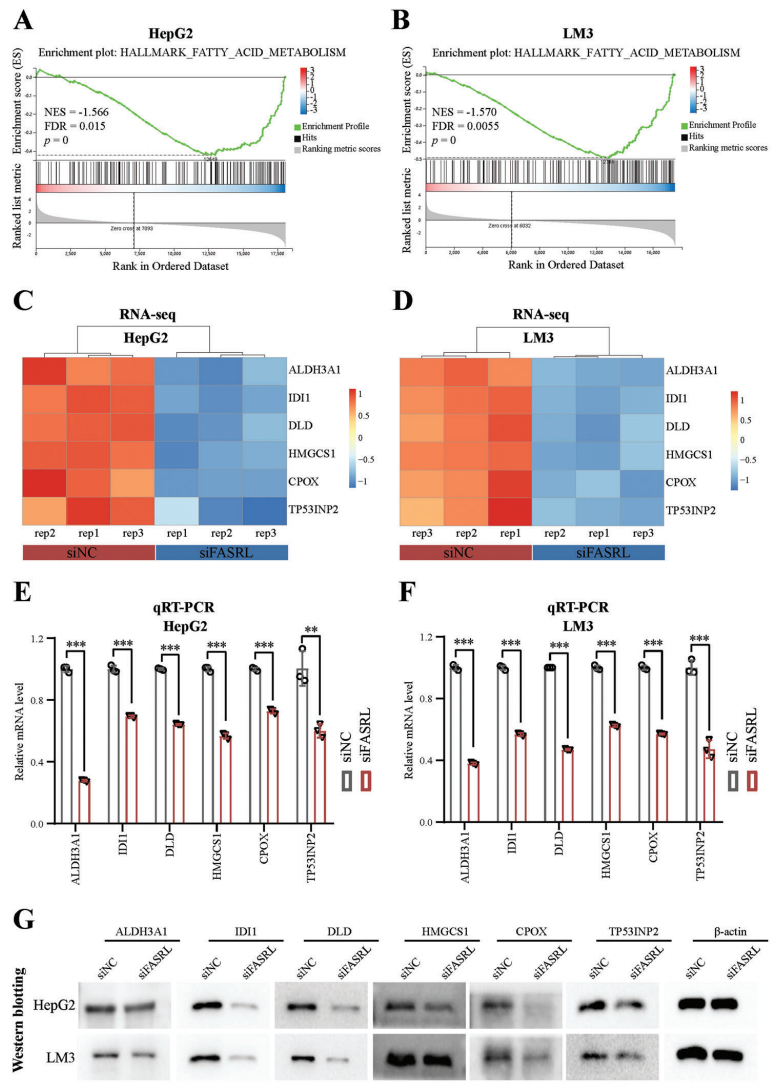
Figure 7 FASRL参与HCC脂肪酸代谢通路[5]。A,B 敲除FASRL表达后表达改变的基因在脂肪酸(FA)代谢通路中富集。C,D qRT-PCR显示FASRL表达下调后,FA代谢通路中表达显著改变的基因表达量热图。E,F FASRL的表达下调后,FA代谢通路中基因的mRNA表达显著下调。G. WB显示FASRL 表达在 HepG2 和 LM3 细胞系中敲低后 FA 代谢途径中基因的蛋白质含量改变。
5、为了进一步探索与FASRL直接相互作用的蛋白质,作者使用RNA Pull Down-MS鉴定与FASRL直接相互作用的RNA结合蛋白,MS结果显示乙酰辅酶A羧化酶1(ACACA)直接与FASRL结合,之后进行RIP-qPCR、FISH、IHC和WB进行进一步验证。先前的研究表明,ACACA (Ser79) 的磷酸化抑制其酶活性并减少 FA 合成,而本研究发现敲低FASRL表达增加了磷酸化ACACA的水平,FASRL与ACACA结合并抑制其磷酸化,从而增加了FA的合成。
6、ACACA是脂质代谢中的重要酶,是控制从头FA生物合成的中心酶。作者用同位素13C6-标记葡萄糖追踪FA合成,证明敲低FASRL抑制了从头FA合成,另外FASRL敲低显著降低了甘油三酯(TG)含量。之后通过油红O染色实验和对脂滴荧光标记进一步验证了在敲低FASRL表达后HCC细胞中的脂滴数量显著减少。
7、综上所述,这项工作发现USF1可以通过SE驱动FASRL表达,FASRL和ACACA之间的相互作用增加FA合成,以促进肿瘤进展(Fig 8)。作者期望未来开发抑制TF USF1或SE的药物来降低FASRL的表达或抑制FASRL与ACACA的结合以减少FA合成,最终达到抑制HCC的目的。
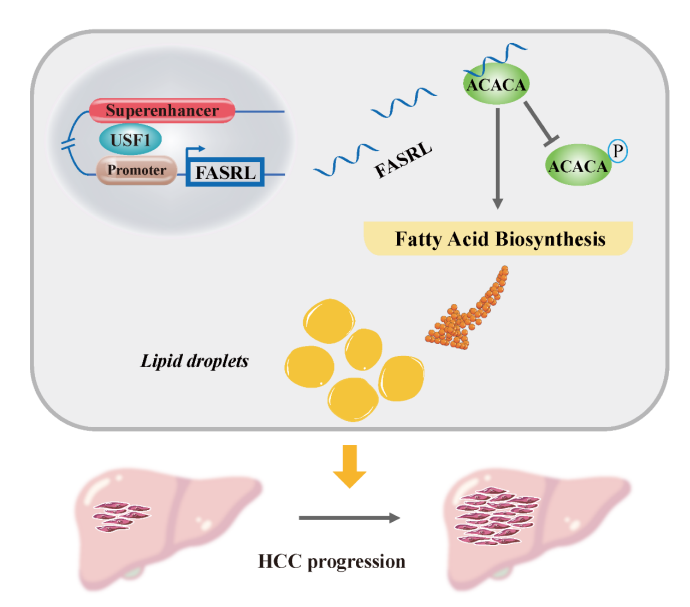
Figure 8 SE-lncRNA FASRL影响肝癌进展机制[5]。
总结以上两篇文章中涉及实验有ChIP-seq、ChIP-qPCR、RNA-seq、RIP-qPCR、RNA Pull down-MS、双荧光素酶报告基因、CoIP-MS、细胞功能实验、动物建模实验等。对于这些实验,武汉金开瑞生物均有大量项目经验,可助力您课题研究,欢迎各位老师咨询!

参考文献:
[1] Zeitlinger J. Seven myths of how transcription factors read the cis-regulatory code. Curr Opin Syst Biol. 2020;23:22-31. doi:10.1016/j.coisb.2020.08.002
[2] Preissl S, Gaulton KJ, Ren B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat Rev Genet. 2023;24(1):21-43. doi:10.1038/s41576-022-00509-1
[3] Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307-319. doi:10.1016/j.cell.2013.03.035
[4] Liang W, Shi C, Hong W, et al. Super-enhancer-driven lncRNA-DAW promotes liver cancer cell proliferation through activation of Wnt/β-catenin pathway. Mol Ther Nucleic Acids. 2021;26:1351-1363. Published 2021 Nov 3. doi:10.1016/j.omtn.2021.10.028
[5] Peng JY, Cai DK, Zeng RL, et al. Upregulation of Superenhancer-Driven LncRNA FASRL by USF1 Promotes De Novo Fatty Acid Biosynthesis to Exacerbate Hepatocellular Carcinoma [published online ahead of print, 2022 Oct 28]. Adv Sci (Weinh). 2022;10(1):e2204711. doi:10.1002/advs.202204711
最新动态
-
07.08
葡萄外泌体“激活细胞清道夫”——SM@G-ELNs协同水凝胶通过MERTK介导的巨噬细胞胞葬作用促进糖尿病伤口愈合(IF=14.3)
-
07.08
三篇顶刊论文,同一个技术选择——BiFC如何破解植物蛋白互作难题?
-
06.24
再登《Cancer Research》!金开瑞双荧光素酶报告系统助力机制研究新突破
-
06.24
登上《Gut》(IF 26.8)!南方医科大学团队揭示葛根类外泌体纳米囊泡靶向肠道细菌治疗类风湿关节炎新机制
-
06.18
亚细胞定位预测与验证结果不一致的原因是什么?
-
06.18
无需抗体、跨物种通用:三篇高水平论文实证DAP-seq如何助力植物转录调控?
-
06.11
打破“单向用药”的局限!IF=11.9《Asian J Pharm Sci》证实:红参外泌体,口服就能双向调控骨代谢
-
06.11
植物外泌体研究前沿:2026年4—5月6项突破性成果系统梳理
-
06.11
IF 26.8!颠覆“越小越好”的认知:南方医科大团队用40微米的柠檬胶囊,让肠屏障主动“开门”抗癌
-
06.11
从零开始研究一个基因,这篇讲透了!
 X
X 






