
导师压箱底的DNA pull down实验全流程拆解!
DNA pull-down技术是一种广泛应用于分子生物学领域的体外实验方法,主要用于鉴定与特定DNA序列相互作用的蛋白质。该技术通过将目标DNA片段(如启动子、增强子或其他调控元件)固定于固相支持物上,进而从细胞核提取物或纯化的蛋白混合物中“沉淀”或“捕获”与之特异性结合的蛋白质。其核心在于利用生物素-链霉亲和素等高效亲和系统,实现对DNA-蛋白复合物的分离与富集。
作为研究基因转录调控、染色质修饰及蛋白-DNA互作的关键手段,DNA pull-down技术能够直接验证蛋白质与特定DNA区域的结合能力,为揭示基因表达调控网络提供实验依据。
一、实验原理
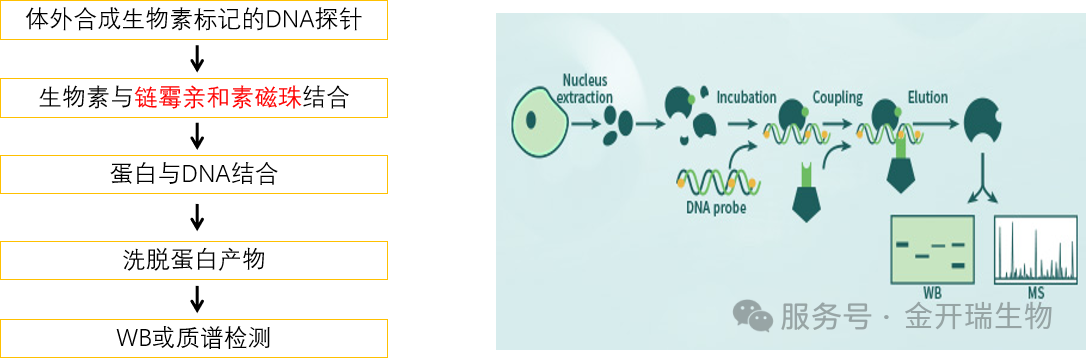
DNA pull down是利用生物素标记的DNA探针,通过与含有目的蛋白的溶液孵育,形成DNA-蛋白质复合物。复合物可以特异性结合磁珠,从而与孵育液中的其他蛋白分离。复合物洗脱后,通过蛋白凝胶电泳实验,检测特定的DNA与蛋白的结合情况。DNA pull-down主要用于目的DNA片段的互作蛋白(如转录因子等)筛选。
二、实验目的
1、解析基因表达调控机制:
这是最主要应用,通过鉴定与启动子、增强子、沉默子等调控区域结合的转录因子、共激活/共抑制复合物,来阐明基因如何被开启或关闭。
2、绘制蛋白质-DNA相互作用图谱:
结合高通量测序(如ChIP-seq后的验证),对基因组上特定区域的结合蛋白进行功能验证和深入分析。
3、疾病机制研究:
研究疾病相关基因突变(尤其是非编码区的突变)如何通过影响蛋白质-DNA结合,导致基因表达失调,从而引发疾病(如癌症、遗传病)。
4、药物靶点发现与验证:
如果某个致病蛋白需要通过结合特定DNA来发挥作用,该技术可用于筛选能干扰此相互作用的小分子化合物。
三、样本准备
1、细胞量:约3*10⁷个细胞,或者3个10cm培养皿,细胞密度达到80-90%,离心后收细胞沉淀,沉淀体积约为80-100μL;
2、动物组织:不少于0.5g,新鲜取材;
3、植物组织:不少于1g,新鲜幼嫩组织;
4、注意:经过药物处理的细胞或组织,需使用PBS冲洗2次后,再收集样品,以免药物对后续实验产生干扰;
5、模板质粒:体积不少于20μL,浓度不低于20ng/μL,冰袋寄送;
6、WB级别抗体大于等于10μL,(仅WB需要)。
四、标准实验流程
一个典型的DNA Pull-Down实验包含以下核心步骤:
1、探针设计与制备
对于研究的启动子靶标区域比较明确,且长度较短的DNA片段,可以直接合成并标记上生物素作为pull down探针;
对于研究的启动子靶标区域没具体预测区域,一般针对基因上游2000 bp序列设计引物,并标记上生物素;然后通过PCR扩增的方式钓取基因上游2000 bp序列,回收纯化后作为DNA pull down的探针。
2、蛋白样本制备
样本前处理:动物组织经PBS清洗后机械匀浆;细胞直接收集;植物组织经液氮研磨成粉末。
裂解与抑制:分别使用相应的冷裂解液(动物IP裂解液或植物蛋白裂解液),以维持蛋白完整性与活性。
破碎与裂解:样本冰浴裂解后,均进行冰浴超声破碎(功率20%,工作/间歇各3秒),持续时间依样本类型调整(动物组织5分钟,细胞2分钟,植物组织10分钟)。
离心收集:所有样本最终经4℃、12000 r/min离心10分钟,取上清即得总蛋白。
(注意蛋白提取整个过程都在冰上操作,减少高温造成的蛋白降解;超声过程中最好不要有气泡产生,减少蛋白降解。总蛋白放在-20℃保存,避免反复冻融,可取出一部分放在4℃保存)。
3、磁珠准备及洗涤
(1)将链酶亲和素磁珠从4℃冰箱取出,上下颠倒多次混匀磁珠储存液,分别取30μL到2个1.5mL EP管中,记为对照组和实验组,置于磁力架上静置1min以分离磁珠,弃上清;
(2)加入500μL 核酸缓冲液,重悬磁珠,置于磁力架上1min,弃上清;
4、磁珠结合DNA
实验组加入100-300pmol带生物素标记的DNA探针,对照组加入等量不带生物素标记的DNA,均用核酸缓冲液补至500μL,静音混合仪上室温孵育2h。磁分离收集上清保存。随后用核酸缓冲液1mL洗涤三次,每次磁分离后弃上清。
5、DNA-磁珠结合蛋白
每管加入300–2000μg提取蛋白及1mM PMSF,用蛋白孵育缓冲液补至1mL,4℃孵育过夜,另取100μL裂解液作为Input组。孵育后磁分离收上清保存。用蛋白孵育缓冲液1mL洗涤五次,注意:洗脱前需室温复温0.5h。
6、洗涤与洗脱
每管加100μL洗脱液,沸水浴8–10min,离心取上清,加入Loading buffer再次煮沸,得Pull-down产物。Input组同样加Loading buffer煮沸,所有样品-20℃保存。注:若样本用于质谱,最后一次洗涤前取1/4磁珠混合液,用PBS洗涤三次后,-20℃保存磁珠。
7、检测与分析
Western Blot分析:如果针对已知蛋白,可将洗脱产物进行SDS-PAGE电泳和Western Blot,使用特定抗体检测目标蛋白是否存在。
质谱分析:如果旨在发现新蛋白,可将洗脱产物进行SDS-PAGE染色、切胶或直接进行液相色谱-串联质谱(LC-MS/MS)分析,鉴定所有下拉蛋白的身份。
五、实验优缺点
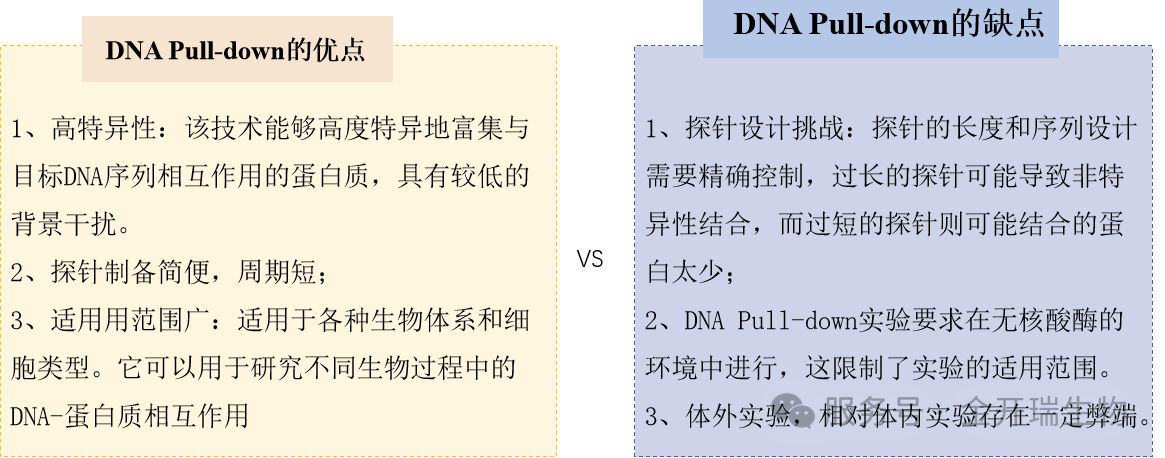
与体内实验的关联:DNA Pull-Down的阳性结果应尽可能与体内功能实验(如ChIP-qPCR、报告基因实验、基因敲低/敲除后的表型分析)相互印证,以确认其生物学相关性。
技术联用:通常作为EMSA(验证结合)、ChIP(验证体内结合)和酵母单杂交(筛选)等技术的补充和验证手段,共同构成DNA-蛋白质互作研究的证据链。
六、如何看结果?
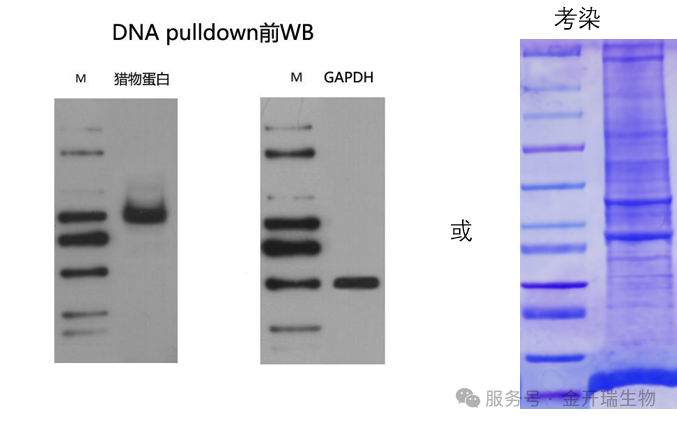
1、在正式实验前,建议先通过WB检测预期的猎物蛋白的本底表达量,如果表达水平太低,pull down后可能检测不到而误判成假阴性结果,建议样本中过表达后在尝试,同时也可以借此判断样本中蛋白是否已发生降解。
2、如果没有WB条件或需求,亦可以通过考染判断蛋白提取质量。
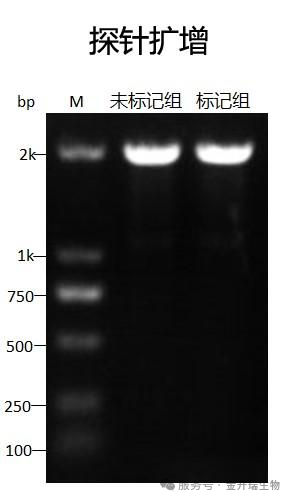
1、扩增好的DNA探针通过凝胶电泳,判断扩增是否完全;
2、注意:未标记的DNA探针作为对照组,生物素标记的DNA 作为实验组。
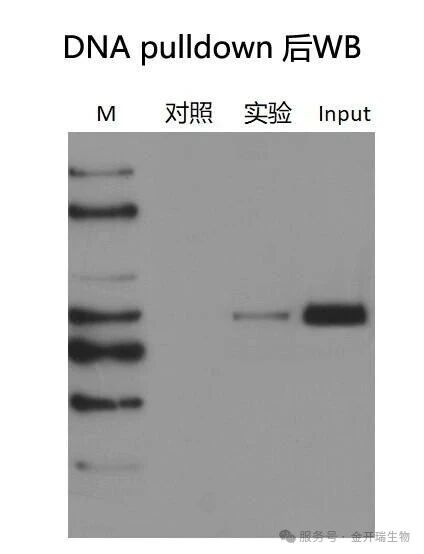
pull down后通过WB检测猎物蛋白,若实验组能检测到猎物蛋白目的条带,而对照组没有,说明该蛋白能与诱饵DNA特异性结合;若都没有检测到,说明该蛋白不能与诱饵DNA结合。

银染通常作为质谱送样上机前的质控,检测pull down后蛋白液的质量,或在知道预期蛋白大小的前提下切胶送质谱。
七、常见问题
1、非特异性结合:蛋白质可能与非特定的DNA序列结合,导致背景信号增加。
尝试优化DNA探针的设计,选择更特异的DNA序列作为探针,以增加结合的特异性。还可以通过引入竞争性DNA或非特异性DNA序列来减少非特异性结合。
2、DNA pull-down实验中,SDS PAGE之后,为什么用银染,而不是考染?
由于洗脱得到的蛋白丰度较低,银染检测的灵敏度高,能够检测到ng级的蛋白质。通过银染,也能够判断蛋白提取的质量,以及Pull Down之后蛋白洗脱的效果。
3、DNA pull-down需要做质谱时,如何选择定量和不定量?
根据自己的实验需要来选择,如果要知道两者之间的差异蛋白(如用质谱比较空白对照与目的蛋白过表达之间的差异蛋白来鉴定与目的蛋白相互作用),就需要定量,如果只是想知道所提供的材料里面的蛋白种类就不需要定量。
4、目标蛋白结合量少或无结合?
保证蛋白活性:使用新鲜核提取物或细胞裂解液,全程冰上操作,加入足量蛋白酶和磷酸酶抑制剂。
优化孵育条件:通常4°C旋转孵育过夜效果最佳,有利于结合平衡并减少蛋白降解。可测试1-2小时室温孵育(针对强相互作用)。
增加探针量:确保探针过量,完全捕获目标蛋白。
5、检测灵敏度低,WB信号弱?
可能是上样量不足,蛋白在洗涤过程中损失。
解决方案:
浓缩样品:对于洗脱液,可用丙酮或TCA沉淀法浓缩蛋白。
直接上样:将磁珠-蛋白复合物直接与上样缓冲液混合煮沸,全部上样,避免洗脱步骤的损失。
使用高灵敏度检测系统:如化学发光底物增强液,或使用荧光二抗配合荧光成像仪。
验证抗体:确保抗体能识别变性(WB)或天然(若用竞争洗脱)状态下的目标蛋白。
最新动态
-
07.08
葡萄外泌体“激活细胞清道夫”——SM@G-ELNs协同水凝胶通过MERTK介导的巨噬细胞胞葬作用促进糖尿病伤口愈合(IF=14.3)
-
07.08
三篇顶刊论文,同一个技术选择——BiFC如何破解植物蛋白互作难题?
-
06.24
再登《Cancer Research》!金开瑞双荧光素酶报告系统助力机制研究新突破
-
06.24
登上《Gut》(IF 26.8)!南方医科大学团队揭示葛根类外泌体纳米囊泡靶向肠道细菌治疗类风湿关节炎新机制
-
06.18
亚细胞定位预测与验证结果不一致的原因是什么?
-
06.18
无需抗体、跨物种通用:三篇高水平论文实证DAP-seq如何助力植物转录调控?
-
06.11
打破“单向用药”的局限!IF=11.9《Asian J Pharm Sci》证实:红参外泌体,口服就能双向调控骨代谢
-
06.11
植物外泌体研究前沿:2026年4—5月6项突破性成果系统梳理
-
06.11
IF 26.8!颠覆“越小越好”的认知:南方医科大团队用40微米的柠檬胶囊,让肠屏障主动“开门”抗癌
-
06.11
从零开始研究一个基因,这篇讲透了!
 X
X 






