
双荧光素酶实验,如何助力客户连发IF>10高分文章?揭秘我们的核心技术与成功案例!
双荧光素酶报告基因实验(Dual-Luciferase Reporter Assay)是研究基因转录调控、信号通路激活以及非编码RNA靶向验证的关键分子生物学技术。该体系基于萤火虫荧光素酶(Firefly Luciferase)和海肾荧光素酶(Renilla Luciferase)两种报告基因,通过共转染至细胞中,分别检测目标启动子或3′UTR的转录活性或靶向调控效应。萤火虫荧光素酶作为实验报告基因,反映目标序列的调控活性;海肾荧光素酶作为内参,校正转染效率与细胞活性差异,确保结果的可靠性与重复性。
该技术具有灵敏度高、线性范围宽、操作便捷、适用性广等优势,已成为研究转录因子与DNA互作、miRNA与靶基因结合、信号通路调控等领域的“金标准”实验手段。
一、主要应用方向
1、验证miRNA与靶基因结合
将靶基因序列构建到萤⽕⾍荧光素酶基因3’区域,同时过表达microRNA。当microRNA与靶基因上特异结合位点结合后,将⼲扰荧光素酶mRNA的翻译或导致其迅速降解,使荧光强度降低;当结合位点被突变后,microRNA⽆法与靶基因结合,因此荧光值⽆明显变化。
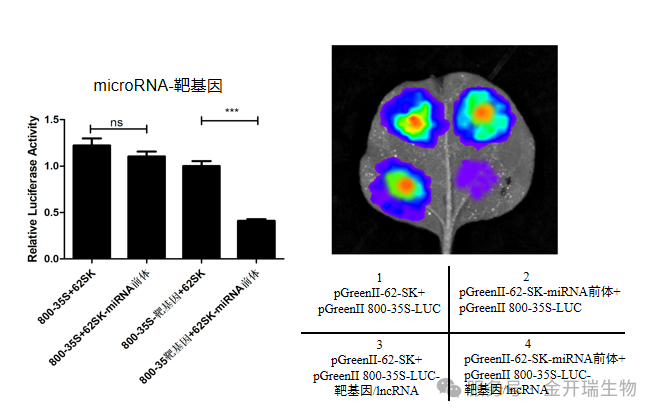
2、验证转录因子与启动子结合
将启动⼦序列构建到萤⽕⾍荧光素酶基因前,同时过表达转录因⼦。当转录因⼦与启动⼦上特异结合位点结合后激活荧光素酶基因转录,使萤⽕⾍荧光素酶得以表达,最终荧光强度上升;当结合位点被突变后,转录因⼦⽆法与启动⼦结合,因此荧光值⽆明显变化。
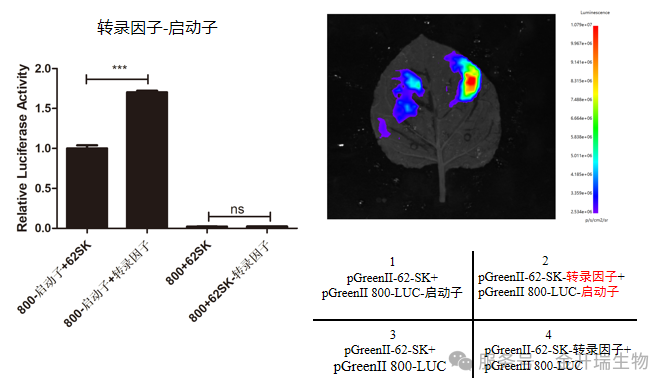
以下为金开瑞与多家科研团队合作发表的高水平研究中,双荧光素酶实验在解析分子机制中的关键应用案例。
案例1:验证肿瘤来源miRNA 对肌肉萎缩相关基因的靶向调控

2025年7月
期刊:Advanced Science (IF=14.1)
研究题目:Pancreatic Cancer-Derived Extracellular Vesicles Enriched with miR-223-5p Promote Skeletal Muscle Wasting Associated with Cachexia
研究背景:胰腺癌来源的胞外囊泡(EVs)可通过递送 miR-223-5p 促进肌肉萎缩,但其直接靶基因及调控方式尚不明确。
双荧光素酶实验设计:
为验证miR-223-5p 是否直接靶向 MAFA 基因的 3′UTR,研究构建了两种报告基因载体:一种携带 MAFA 3′UTR 的野生型序列,另一种则在预测的 miR-223-5p 结合位点引入突变。将报告基因载体分别与 miR-223-5p 模拟物(mimics)或对照共转染 C2C12 成肌细胞。
关键结果:
过表达miR-223-5p 可显著抑制野生型 MAFA 3′UTR 的报告基因活性,突变miR-223-5p 的结合位点后,其抑制效应完全消除。
科学意义:
该结果在分子水平直接证实了miR-223-5p 通过结合 MAFA mRNA 的 3′UTR 抑制其表达,阐明了胰管腺癌(PDAC)来源EVs 通过 miRNA 调控肌肉基因表达、导致萎缩的精确路径,为开发靶向治疗策略提供了新靶点。
案例2:阐明缺血性卒中神经元损伤的miRNA调控网络

发表时间:年7月
期刊:Journal of Advanced Research(IF:13.0)
研究题目:Endothelial cells secrete small extracellular vesicles to promote neuron endoplasmic reticulum stress injury via miR-146a-5p/Eif4g2 axis in ischemic stroke
研究背景:
内皮细胞来源的小细胞外囊泡通过传递miR-146a-5p促进神经元内质网应激损伤,但其直接靶基因及作用机制不明。
双荧光素酶实验设计:
为验证miR-146a-5p是否直接靶向Eif4g2的3'UTR,研究者构建了:野生型Eif4g2 3'UTR报告载体;突变型Eif4g2 3'UTR报告载体(突变miR-146a-5p结合位点)
实验结果:
miR-146a-5p模拟物显著抑制野生型Eif4g2 3'UTR的荧光素酶活性,突变结合位点后,miR-146a-5p的抑制效应消失,该结果在体内外模型中得到一致验证。
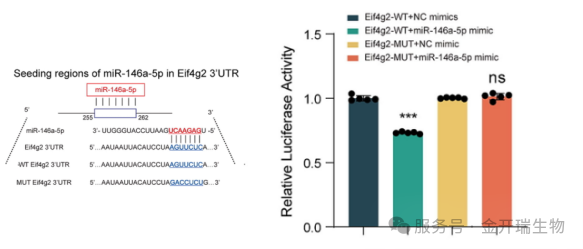
科学意义:
该实验证实Eif4g2是miR-146a-5p的直接靶基因,揭示了内皮细胞-神经元通过miRNA-靶轴调控内质网应激的新机制,为卒中治疗提供了新靶点。
案例3:揭示环境毒物诱导神经毒性的转录调控机制

发表时间:2024年9月
期刊:Journal of Hazardous Materials (IF=12.2)
研究题目:Targeting m⁶A mRNA demethylase FTO alleviates manganese-induced cognitive memory deficits in mice
研究背景:锰(Mn)暴露可导致学习记忆损伤,但其表观遗传调控机制不明。研究发现,Mn 通过影响转录因子 SOX2 的磷酸化,下调去甲基化酶 FTO 的表达,进而影响突触可塑性相关基因的 m⁶A 修饰与稳定性。
双荧光素酶实验设计:
为了探索Mn如何下调海马中FTO表达的机制,我们使用人和小鼠的JASPAR 、TFDB和GTRD 数据库来选择潜在的转录因子,确定了四种转录因子,SOX 2、GABPA、STAT 3和RUNX 1作为候选物,我们将FTO启动子克隆到编码荧光素酶的pGL 3基本载体中,然后将构建体与转录因子质粒和pRL-TK质粒一起转染到HEK 293 T细胞中,随后进行荧光素酶报告基因测定。
关键结果:
只有SOX2 可显著激活 FTO 全长启动子的报告基因活性,截短实验锁定其核心结合区域位于启动子−700bp 至−400 bp 区间。点突变该区域内预测的 SOX2 结合序列(“aaacaatagac” → “gggtgccgagt”)后,相对荧光素酶活性明显降低。
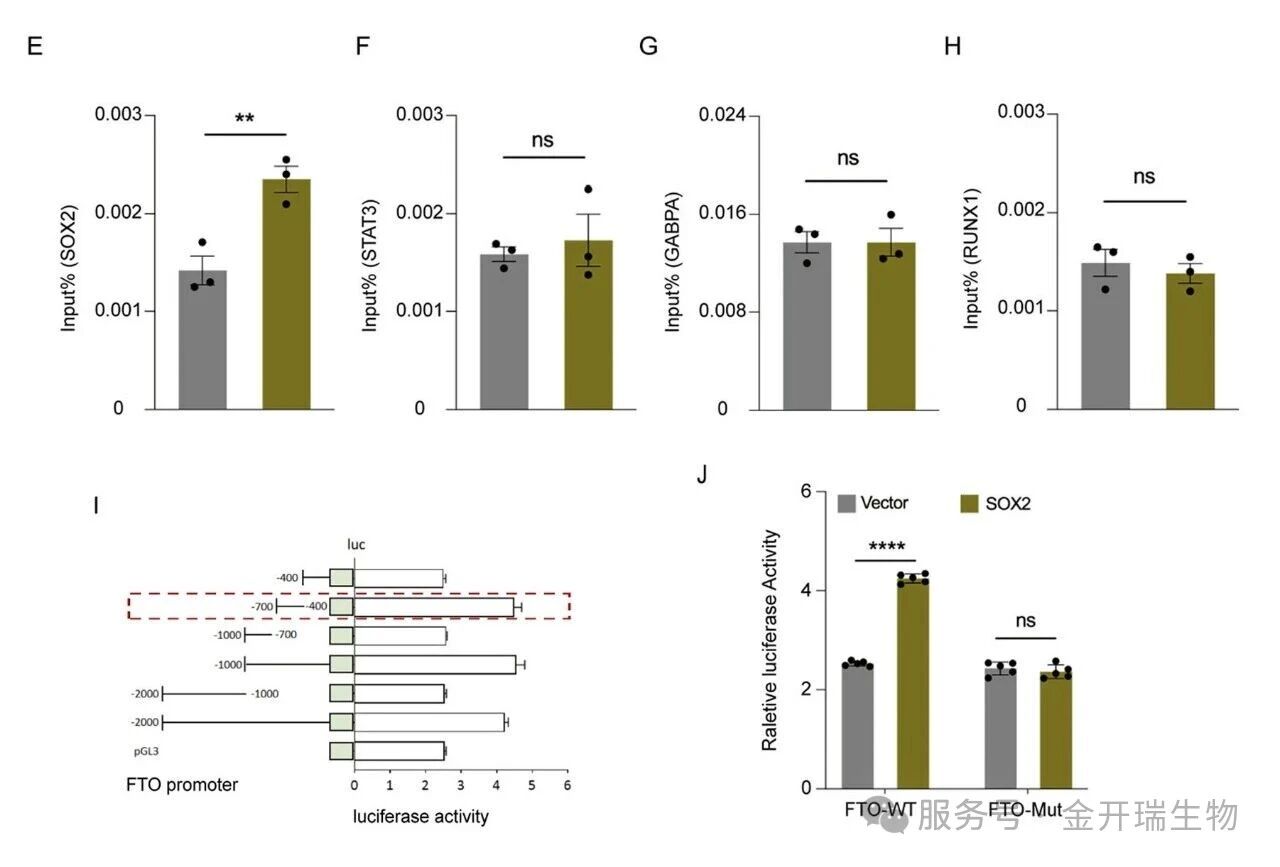
图注:(E-H)SOX 2、STAT 3、GABPA、RUNX 1和FTO双荧光素酶报告定量结果;(I,J)将携带不同截短的纤连蛋白(F-N)启动子的pGL 3-basic载体与pRL-TK共转染入HEK 293 T细胞中。
科学意义:
该实验直接证明了SOX2 作为转录因子直接结合并激活 FTO 启动子,明确了 Mn 通过抑制 SOX2 磷酸化进而下调 FTO 表达的转录层面机制,为理解 Mn 神经毒性的上游调控提供了关键证据。
案例4:揭示膀胱癌化疗耐药新机制

发表时间:2025年7月
期刊:Research (IF=10.7)
研究题目:Combinational Analysis of Metabolomic and O-GlcNAcylation Omics Reveals the HBP Metabolic Regulation of Chemoresistance via GFPT1/NR3C1 O-GlcNAcylation/GPX4 Axis
研究背景:
膀胱癌化疗耐药是临床治疗难点,研究发现NR3C1通过O-GlcNAc修饰增强其转录活性,进而调控下游基因GPX4表达,抑制铁死亡,促进耐药。
双荧光素酶实验设计:
为验证NR3C1是否直接结合GPX4启动子并调控其转录,研究者构建了:野生型GPX4启动子报告载体,突变型GPX4启动子报告载体(突变NR3C1结合位点“GAGACCAGCCTGACCAAC”)
实验结果:
NR3C1过表达显著增强野生型GPX4启动子的荧光素酶活性,突变NR3C1结合位点后,荧光素酶活性显著降低,O-GlcNAcylation位点的突变显著减弱了NR3C1介导的GPX4转录激活。
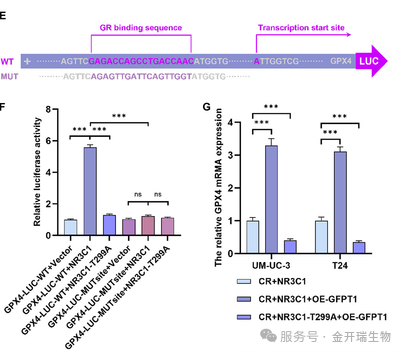
图注:(E) NR3C1在GPX4启动子上的结合序列示意图。(F) 双荧光素酶报告基因检测分析NR3C1在GPX4启动子上的结合位点特异性;检测T299A突变对荧光素酶活性的影响;以GPX4-LUC-WT + Vector组的荧光素酶活性水平为基准进行标准化;采用单因素方差分析及T检验进行统计分析(每组n=4)
科学意义:
该实验直接证实NR3C1通过特异性结合GPX4启动子增强其转录,揭示了O-GlcNAcylation修饰通过增强转录因子活性促进化疗耐药的分子机制。
案例5:揭示心脏移植后心肌缺血再灌注损伤新机制

发表时间:年9月
期刊:Advanced Science(IF:14.1)
研究题目:Inhibition of Macrophage ARID3A Alleviates Myocardial Ischemia-Reperfusion Injury After Heart Transplantation by Reducing THBS1/CD47 Signaling-Mediated Neutrophil Extracellular Traps Formation
研究背景:
心脏移植后,M1型巨噬细胞中ARID3A转录因子上调,可能通过激活THBS1基因表达,进而促进中性粒细胞NETs形成,加重心肌损伤。需验证ARID3A对THBS1的直接转录调控作用。
双荧光素酶实验设计:
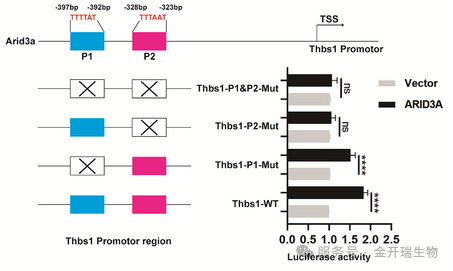
预测与设计:通过JASPAR数据库预测ARID3A在THBS1启动子上的两个潜在结合位点(P1, P2),并据此构建:野生型(WT)THBS1启动子荧光素酶报告载体。单位点(Mut P1, Mut P2)及双位点(Mut P1&P2)突变型报告载体。
功能验证:将上述报告载体与ARID3A过表达载体共转染293T细胞,检测荧光素酶活性变化。
关键结果:
ARID3A过表达可显著激活野生型THBS1启动子。
仅当P2位点突变时,ARID3A的激活作用完全丧失,证明P2是ARID3A的功能性结合位点。
ChIP实验进一步证实ARID3A蛋白直接结合于THBS1启动子的P2区域。
双荧光素酶报告器检测THBS1启动子活性。
科学意义:
该实验直接证实ARID3A通过特异性结合THBS1启动子的P2位点正向调控其转录,从而确立了ARID3A-THBS1轴在驱动巨噬细胞介导心肌损伤中的核心分子机制,为靶向干预提供了关键依据。
最新动态
-
07.08
葡萄外泌体“激活细胞清道夫”——SM@G-ELNs协同水凝胶通过MERTK介导的巨噬细胞胞葬作用促进糖尿病伤口愈合(IF=14.3)
-
07.08
三篇顶刊论文,同一个技术选择——BiFC如何破解植物蛋白互作难题?
-
06.24
再登《Cancer Research》!金开瑞双荧光素酶报告系统助力机制研究新突破
-
06.24
登上《Gut》(IF 26.8)!南方医科大学团队揭示葛根类外泌体纳米囊泡靶向肠道细菌治疗类风湿关节炎新机制
-
06.18
亚细胞定位预测与验证结果不一致的原因是什么?
-
06.18
无需抗体、跨物种通用:三篇高水平论文实证DAP-seq如何助力植物转录调控?
-
06.11
打破“单向用药”的局限!IF=11.9《Asian J Pharm Sci》证实:红参外泌体,口服就能双向调控骨代谢
-
06.11
植物外泌体研究前沿:2026年4—5月6项突破性成果系统梳理
-
06.11
IF 26.8!颠覆“越小越好”的认知:南方医科大团队用40微米的柠檬胶囊,让肠屏障主动“开门”抗癌
-
06.11
从零开始研究一个基因,这篇讲透了!
 X
X 






