
怎么通过ChIP-seq结果分析转录因子的结合基序与结合位点分布?
ChIP-seq染色质免疫共沉淀结合高通量测序是研究转录因子在全基因组上结合规律最核心的技术。简单说,就是把细胞里的DNA和蛋白交联,用转录因子特异性抗体把目的蛋白拉下来,顺带把它结合的DNA片段富集,再做测序。最终我们拿到两大核心分析方向:一是转录因子的保守结合基序(motif),也就是它最喜欢识别、绑定的DNA短序列;二是结合位点在基因组上的分布特征,比如集中在启动子、增强子、基因间区、内含子,以及在TSS上下游的位置偏好。
很多人跑完ChIP-seq上机、拿到原始数据后,只会看峰图、找几个靶基因,不知道怎么系统分析结合基序和位点分布。本文从数据分析逻辑、实操步骤、结果解读、生物学意义四个层面,完整讲透怎么从ChIP-seq结果里挖转录因子的结合规律。
一、ChIP-seq前期基础分析:做基序和分布分析的前提
想分析基序和位点分布,不能直接拿原始测序reads就开始做,必须先完成标准上游分析,这是所有后续分析的基础。
第一步:原始测序数据质控。对fastq文件做质量过滤,去掉低质量reads、接头污染、过短序列,保证测序数据干净可靠。
第二步:序列比对。把干净的reads比对到参考基因组,常用软件Bowtie、BWA,把每条测序片段定位到染色体具体位置。
第三步:峰鉴定(Peak calling)。这是最关键一步。转录因子结合DNA的区域,reads富集度远高于背景噪音,通过MACS2等软件统计显著性,筛选出显著富集的结合峰(Peak)。每一个 Peak 就代表转录因子在基因组上的一个真实结合位点。
只有拿到高质量、去冗余、显著性达标的Peak文件,才能往下做结合基序分析和基因组分布分析。如果Peak假阳性多、背景噪音大,后面基序会乱、分布比例完全失真。
二、ChIP-seq分析转录因子结合基序(Motif)完整逻辑
1. 什么是转录因子结合基序
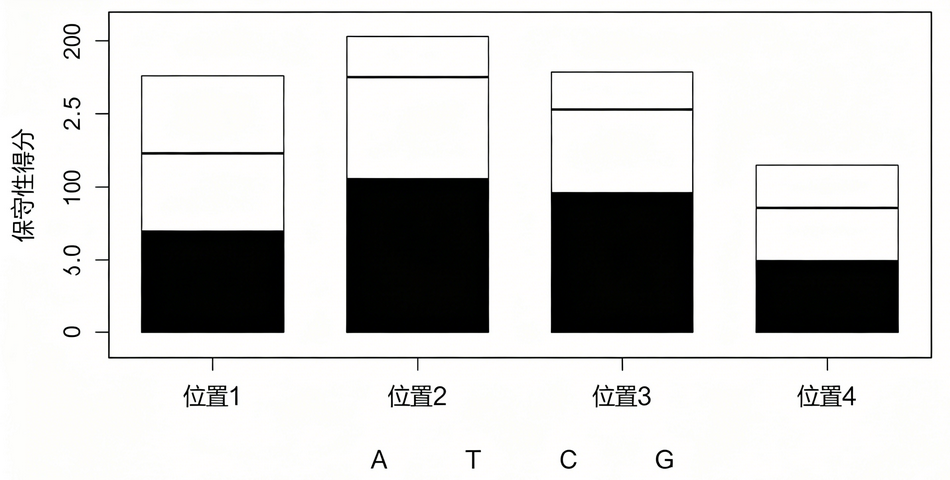
转录因子不是随机乱绑DNA,它有固定的DNA识别序列,一般长度6–20bp,具有保守的碱基排列模式,这就是Motif 结合基序。
比如常见的 AP-1、NF-κB、p53都有自己经典的保守基序。ChIP-seq的核心价值之一,就是不依赖已知文献,从头挖掘该转录因子全新的、最偏好的结合序列。
2. 基序分析两种主流方式
(1)从头预测De novo Motif
把所有ChIP-seq的Peak中心上下游截取固定长度序列,一般取200bp或500bp,用MEME、Homer、STREME等软件,无参考、从头挖掘富集度最高、最保守的 DNA 序列模式。
这种方式最大优势:
可以找到全新的未知基序,也能找出该转录因子第一偏好、第二偏好的多个 motif,能发现它和其他转录因子的共结合模式。
(2)已知基序匹配注释
把Peak序列和数据库里已知的转录因子Motif库做比对,比如JASPAR、HOCOMOCO数据库,看富集到的序列和哪些已知转录因子基序高度相似。
用来验证:你研究的转录因子,是不是确实结合经典保守序列;同时还能看它常和哪些其他转录因子共用结合位点,形成转录调控网络。
3. 怎么判断基序结果是否可靠
第一:富集显著性E-value、P值越小越可靠;
第二:该Motif在所有Peak中出现比例越高,说明特异性越强;
第三:Motif集中出现在 Peak中心位置,说明是真实特异性结合,不是随机背景序列;
第四:和已发表同物种、同转录因子的经典Motif做比对,相似度高说明结果可信。
4. 基序结果能解读哪些生物学信息
确定转录因子核心保守识别序列,明确它偏好结合AT富集区还是GC富集区;
发现次要结合基序,说明该转录因子可以多种模式结合DNA;
挖掘共结合转录因子,看它常和谁协同调控基因;
区分特异性结合和非特异性背景结合,过滤假阳性Peak。
三、ChIP-seq分析转录因子结合位点的基因组分布
拿到全部显著Peak后,第二个核心分析就是统计所有结合位点在基因组功能区域的分布规律。基因组一般划分为:启动子区、TSS附近、5'UTR、3'UTR、外显子、内含子、基因间区、增强子区域。不同类型转录因子分布规律完全不一样:
通用转录因子、管家相关因子:大量集中在启动子、TSS附近;
信号通路转录因子、发育调控因子:大量分布在增强子、基因间区、内含子;
部分转录因子既有启动子结合,又有远端增强子结合,同时参与近端和远端调控。
1. 位点分布分析具体怎么做
第一步:Peak注释。用ChIPseeker、Homer、ANNOVAR等工具,把每一个 Peak 定位到对应的基因组结构区域,注释属于启动子、内含子、基因间区还是外显子。
第二步:统计比例。统计所有Peak在各个区域的数量占比,画饼图、柱状图,直观看到偏好分布。
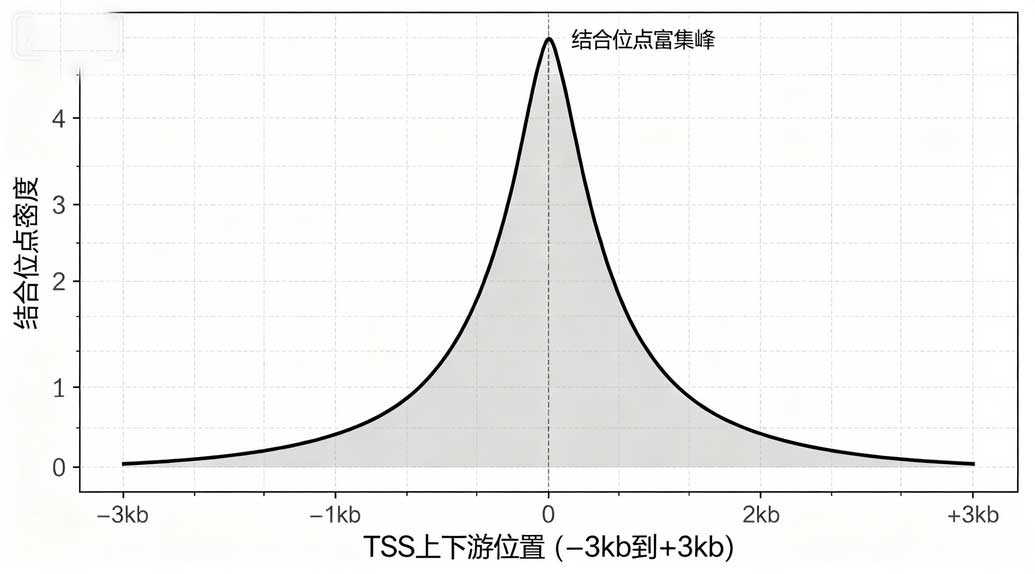
第三步:TSS上下游分布分析。以基因转录起始位点TSS为原点,统计Peak在TSS上下游±1kb、±3kb、±5kb的富集密度,画出位点密度分布图,看转录因子最喜欢结合在 TSS 上游多远的位置。
第四步:染色体分布统计。看结合位点在各条染色体上的分布是否均匀,有没有染色体偏好性。
第五步:与增强子、超级增强子重叠分析。把 Peak 和已知增强子区域比对,看转录因子是否大量富集在增强子区,判断它是近端启动子调控还是远端增强子调控。
2. 分布结果怎么解读生物学意义
如果大量Peak集中在启动子TSS附近
说明该转录因子主要直接调控基因转录起始,偏向管家基因、基础表达调控。
如果大量Peak落在基因间区、内含子区
说明主要通过远端增强子发挥作用,远距离调控基因表达,多见于发育、分化、肿瘤相关转录因子。
TSS分布峰位置靠前(上游200–500bp富集最高)是典型的启动子结合特征;
如果远离TSS、在±1–3 kb富集,属于近端调控区;
更远则属于典型增强子区域结合。
与超级增强子高度重叠
说明该转录因子是细胞身份关键调控因子,控制细胞分化、命运决定。
四、结合基序与位点分布联合分析(高阶逻辑)
单独看基序、单独看分布都不够,真正专业的分析是两者结合。
看不同基因组区域的Peak,是否共享同一个核心Motif
如果启动子区、增强子区的Peak都富集同一个保守基序,说明该转录因子无论近端还是远端,都靠同一套序列模式识别 DNA。
不同 Motif 对应不同基因组分布
有的次要Motif更多出现在增强子区,核心Motif更多出现在启动子区,说明不同结合模式参与不同调控通路。
基序中心位置 + Peak 富集峰形联合判断
真实特异性结合:Motif严格落在Peak最高点中心;
随机非特异结合:Motif散乱分布,无中心聚集。
结合靶基因功能富集
把有结合位点的靶基因做GO、KEGG富集,结合分布特征:启动子结合多的基因偏向代谢、基础功能;
增强子结合多的基因偏向信号通路、发育、肿瘤通路。
五、常见分析误区与避坑要点
不做Peak质控直接分析Motif
背景噪音、假Peak太多,会挖出来一堆杂乱无意义的motif,完全不可信。
只看已知Motif,不做De novo从头预测
错过新的结合模式、错过共结合因子,分析流于表面。
只统计分布比例,不做TSS密度分析
看不出转录因子精确偏好的调控位置,结论太笼统。
不区分特异性结合与背景结合
把大量随机Peak纳入分析,导致分布比例、Motif富集全部失真。
忽略生物学重复
ChIP-seq必须重复,只做单样本Peak分析,基序和分布都不可靠。
通过ChIP-seq结果分析转录因子,核心就是两大块:结合基序分析和位点基因组分布分析。
结合基序分析,依靠 Peak 序列从头预测和已知数据库比对,能找到转录因子保守识别序列、主次偏好motif、以及协同结合的其他转录因子,从分子层面解释它怎么特异性识别DNA。
位点分布分析,通过Peak注释、功能区域统计、TSS分布密度、增强子重叠分析,能判断转录因子主要依赖启动子近端调控还是增强子远端调控,揭示它在细胞生理、发育、疾病中的调控模式。
把基序特征和位点分布联合解读,再结合靶基因功能富集,就能从ChIP-seq数据完整讲清楚:这个转录因子识别什么序列、喜欢绑在基因组什么位置、调控哪些通路、发挥什么生物学功能。整套分析逻辑清晰、流程固定,是转录组、表观遗传、分子生物学研究必备的标准分析思路。
最新动态
-
05.27
DAP‑Seq实验如何设置对照、保证重复性与数据可靠性?
-
05.27
DAP‑Seq与ChIP‑Seq、CUT&Tag、ATAC‑Seq的关键区别与适用场景?
-
05.26
定制ELISA试剂盒的灵敏度、特异性、重复性等关键性能如何保证?
-
05.11
怎么通过ChIP-seq结果分析转录因子的结合基序与结合位点分布?
-
04.27
双分子荧光互补(BiFC)与FRET的核心区别是什么?
-
04.27
外泌体研究方案中的样本来源与实验模型如何设计?
-
04.24
细胞迁移及侵袭实验攻略
-
04.24
等温量热滴定曲线出现正负峰的原因是什么?
-
04.23
EMSA实验中,细胞核蛋白提取质量对结果影响极大,如何保证蛋白的完整性与结合活性?
-
04.23
免疫荧光检测中常见的非特异性荧光有哪些原因?如何减少或避免?
 X
X 






