
ChIP-seq技术在转录因子与组蛋白修饰研究中的核心应用与进展
染色质免疫共沉淀测序(ChIP-seq)自2007年问世以来,已彻底改变了表观遗传学和转录调控领域的研究格局。作为连接基因组序列与功能调控的关键桥梁,ChIP-seq技术能够以全基因组分辨率精准定位蛋白质(如转录因子)或特定修饰组蛋白在染色质上的结合位点。截至2026年,随着单细胞技术的成熟和长读长测序的普及,ChIP-seq已从单纯的“地图绘制”工具,演变为解析复杂基因调控网络、疾病发生机制及细胞命运决定的核心手段。本文将深入探讨ChIP-seq在转录因子结合位点鉴定及组蛋白修饰图谱构建中的应用原理、技术挑战及最新研究进展。
1、技术原理与工作流程
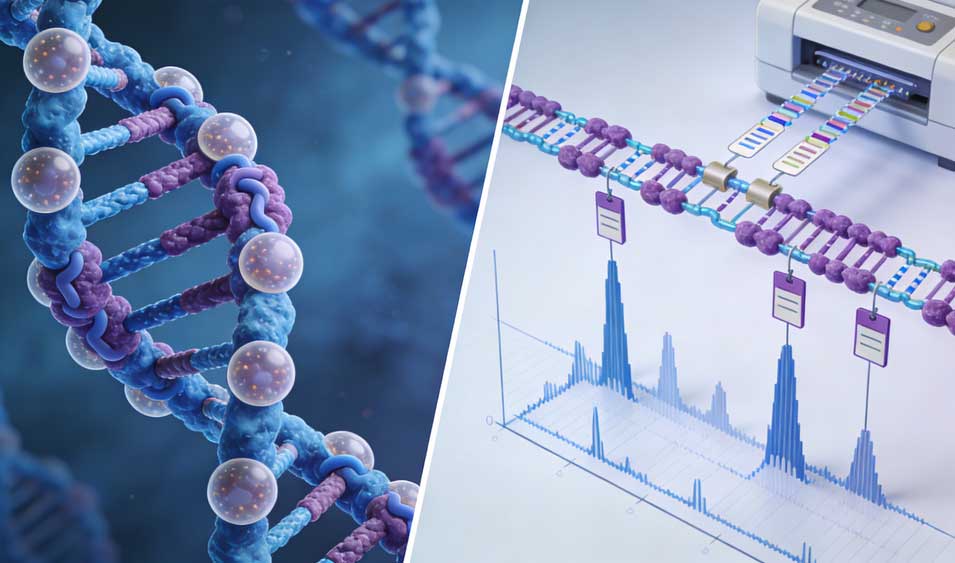
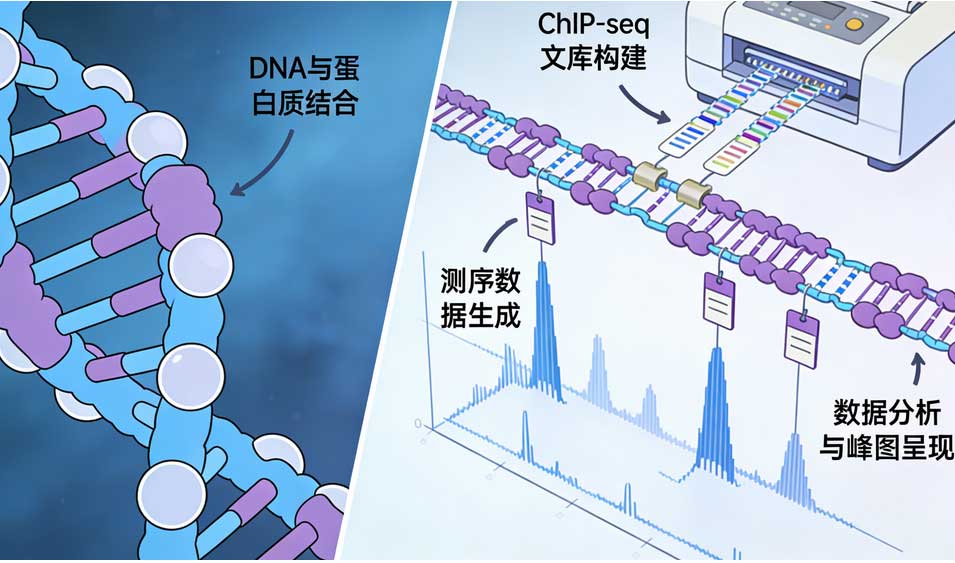
ChIP-seq的核心逻辑在于“富集”与“定位”。其基本流程包括:首先利用甲醛等交联剂将细胞内的蛋白质与DNA共价交联,固定瞬时的相互作用;随后通过超声破碎或酶切将染色质打断成200-500bp的片段;接着利用高度特异性的抗体免疫沉淀目标蛋白(转录因子或特定修饰的组蛋白),从而富集与之结合的DNA片段;最后解交联、纯化DNA并构建文库进行高通量测序。
与早期的ChIP-chip技术相比,ChIP-seq具有无探针限制、动态范围广、分辨率高(可达单碱基水平)等显著优势。对于转录因子研究,由于结合位点通常较窄(峰宽约100-200bp),需要较高的测序深度(通常2000万-4000万读数)以区分特异性结合与背景噪音;而对于组蛋白修饰,由于修饰区域往往较宽(如H3K27me3覆盖整个基因沉默区域,峰宽可达数kb),测序策略和数据分析算法则需相应调整。
2、转录因子结合位点的精准图谱
转录因子(Transcription Factors,TFs)是基因表达的直接调控者,它们通过识别特定的DNA序列基序(Motif)来启动或抑制转录。ChIP-seq是绘制全基因组转录因子结合图谱的金标准。
在基础研究中,ChIP-seq揭示了转录因子结合的复杂性。例如,著名的先锋转录因子(Pioneer Factors)如FOXA1,能够在致密的异染色质区域结合并打开染色质,为其他转录因子的招募铺平道路。通过对比不同细胞类型或刺激条件下的ChIP-seq数据,研究者发现转录因子的结合具有高度的细胞特异性和动态性。同一转录因子在不同细胞中可能结合完全不同的基因组位点,这取决于局部的染色质可及性及协同因子的存在。
在疾病机制研究中,转录因子ChIP-seq数据揭示了致癌驱动因子的异常调控网络。例如,在白血病研究中,RUNX1或MYC的异常结合位点图谱帮助科学家识别了关键的下游靶基因,这些靶基因往往涉及细胞周期失控或分化阻滞。此外,结合全基因组关联分析(GWAS)数据,ChIP-seq还能解释非编码区疾病风险变异的功能机制:许多位于增强子区域的单核苷酸多态性(SNP)恰恰破坏了转录因子的结合基序,从而导致基因表达失调。
3、组蛋白修饰:染色质状态的“语言”
如果说转录因子是基因的“开关”,那么组蛋白修饰则是决定染色质状态和基因活性的“语言”。不同的组蛋白尾端修饰(如甲基化、乙酰化、磷酸化等)对应着截然不同的生物学功能。ChIP-seq通过针对特定修饰抗体的富集,能够绘制出全基因组的“表观遗传景观”。
常见的活性标记包括H3K4me3(主要富集于活跃基因的启动子区)、H3K27ac(标记活跃增强子和启动子)以及H3K36me3(标记正在转录的基因体)。抑制性标记则主要包括H3K27me3(由PRC2复合物介导,导致兼性异染色质形成和基因沉默)和H3K9me3(构成组成型异染色质)。
通过整合多种组蛋白修饰的ChIP-seq数据,研究者定义了“染色质状态”(Chromatin States)。例如,ENCODE项目和Roadmap Epigenomics项目利用隐马尔可夫模型(HMM),将基因组划分为启动子、增强子、转录区、抑制区等十余种状态。这种多维度的图谱不仅预测了基因的表达水平,还揭示了增强子-启动子的远程互作潜力。在发育生物学中,H3K27me3的动态变化被证明是细胞命运决定的关键,它像一种“分子记忆”,在细胞分裂中维持特定基因集的沉默状态,确保干细胞分化的方向性。
4、技术挑战与2026年的新进展
尽管ChIP-seq技术已相对成熟,但在实际应用中仍面临诸多挑战。首先是抗体质量,抗体的特异性和亲和力直接决定实验成败,非特异性结合会导致大量假阳性。其次是输入样本量的限制,传统ChIP-seq通常需要百万级细胞,难以应用于稀有细胞类型或临床微量样本。
5、针对这些问题,近年来技术取得了突破性进展:
微量与单细胞ChIP-seq:基于微流控技术和改进的文库构建方法(如scChIP-seq,nano-ChIP),现在仅需数千甚至数百个细胞即可完成高质量测序。这使得在肿瘤微环境、早期胚胎发育等异质性极高的样本中解析表观遗传特征成为可能。
长读长测序的结合:引入PacBio或Nanopore长读长测序技术,使得ChIP-seq不仅能定位结合位点,还能在同一条DNA分子上同时检测多个修饰或结合事件(单倍型特异性分析),解决了短读长测序在重复区域和等位基因特异性分析上的局限。
多组学整合:当前的研究趋势不再局限于单一的ChIP-seq,而是将其与ATAC-seq(染色质开放性)、RNA-seq(转录组)及Hi-C(三维基因组)数据进行深度整合。这种多维视角能够构建从染色质开放、转录因子结合、增强子激活到基因转录的完整因果链条。
6、数据分析与未来展望
ChIP-seq的数据分析流程已高度标准化,包括比对、去重、峰检测(Peak Calling)、基序分析及差异结合分析等。然而,随着数据量的爆炸式增长,利用人工智能和深度学习模型挖掘数据背后的规律成为新热点。例如,利用卷积神经网络(CNN)预测转录因子结合序列,或基于现有的表观遗传数据预测未知细胞类型的染色质状态。
展望未来,ChIP-seq技术将继续向更高灵敏度、更高通量和更低成本方向发展。随着空间转录组学的兴起,未来的“空间ChIP-seq”或许能在组织原位直接解析蛋白质-DNA相互作用的空间分布,这将极大地推动我们对器官发育、肿瘤浸润及神经环路功能的理解。总之,作为表观遗传学研究的基石,ChIP-seq在解码生命调控密码的征途中,仍将扮演不可或缺的角色。
最新动态
-
07.27
外泌体miRNA测序研究全套实验步骤包含哪些?
-
07.27
检测外泌体CD9、CD63蛋白的实验操作流程是什么?
-
07.27
植物外泌体和人源干细胞外泌体有什么核心差异?
-
07.27
如何设计完整的外泌体miRNA研究实验方案?
-
07.27
外泌体标志物CD9、CD63有哪些常用检测方法?
-
07.10
MST实验只需要微量样品,完整检测一组结合梯度样本的实验周期大概多长?
-
07.10
MST能否直接检测膜蛋白、不稳定蛋白复合物,对蛋白样品纯度与浓度有什么要求?
-
07.10
MST实验时样本荧光标记效率低、信号波动大,该如何优化样本与实验条件?
-
07.10
MST微量热泳动相比SPR、ITC,检测蛋白小分子亲和力有哪些突出优势与局限?
-
07.10
Halo Pull-down全套外包(载体构建+下拉实验+LC-MS/MS质谱鉴定)的报价主要受哪些因素影响?
 X
X 






