
史上最全的ATAC-seq实验技术详解
ATAC-seq(Assay for Transposase-Accessible Chromatin using sequencing)是一种通过高通量测序技术检测染色质开放区域的方法。使用改造的 Tn5转座酶(携带测序接头),靶向插入基因组中开放的染色质区域(即未被核小体紧密包裹的DNA),同时完成DNA的片段化和测序接头的标记,将经转座酶处理的DNA片段通过PCR扩增,构建测序文库并进行高通量测序。
ATAC-seq通过绘制全基因组染色质可及性图谱,揭示细胞类型特异的表观遗传调控机制,目前已广泛应用于发育生物学、疾病研究(如癌症异质性)和药物靶点筛选等领域。
一、技术应用
1.初步判断所研究的课题是否涉及表观遗传调控机制
2.结合基序分析,识别参与基因调控的转录因子
3.识别转录因子调控的靶基因和功能元件
4.与超级增强子鉴定联合分析,明确活性超级增强子的范围
5.更好地了解药物或疾病的基因调控和细胞应答机制
6.识别驱动细胞命运、疾病或应答相关的转录因子
二、技术优势
1、高灵敏度: ATAC-seq可以检测到基因组中极少量的可及染色质区域,使其在样本资源有限的情况下也能获得有意义的结果。
2、低样本需求: 由于ATAC-seq需要的细胞数较少,特别是与其他染色质可及性测序技术相比,可在稀有细胞样本中应用。
3、高通量测序: 使用高通量测序技术,ATAC-seq能够同时分析数百万个片段,提供全面的基因组范围数据。
4、直接检测开放染色质: 通过标记开放染色质区域,ATAC-seq直接反映了基因组的可及性,而不需要附加步骤。
5、快速实验流程: 相比传统的染色质可及性测序方法,ATAC-seq的实验流程简化,减少了样本处理时间。
6、较少的偏差: 由于ATAC-seq使用转座酶进行标记,相对于其他方法,它具有较少的偏差,更好地捕获染色质区域的可及性。
7、分辨率高: ATAC-seq可以提供较高的基因组分辨率,更精确地标记和定位开放染色质区域。
8、可提供与其他组学联合分析思路;提供个性化分析方案。
三、ATAC-seq实验流程

01.细胞核提取
收集目标细胞(新鲜或快速冷冻,确保细胞活性不低于90%,细胞活性至关重要!死细胞会导致背景噪音)。
细胞核提取:将细胞或组织样本加入裂解液,冰上孵育使细胞膜通透,然后离心去除上清,加入PBS重悬细胞核。
细胞核计数:取适量细胞核悬液,加入台盼蓝溶液染色,用血球计数板在显微镜下计数,计算细胞核浓度。
02.转座酶片段化
配置反应体系,将透化后的细胞核与预加载了接头的Tn5转座酶混合。在适宜温度(通常37°C)下孵育(30-60分钟)。Tn5转座酶优先结合并插入到染色质结构开放、无核小体占据或核小体疏松的区域。在插入位点两侧切割DNA双链。同时将预加载的测序接头连接到被切割的DNA片段两端。
03.DNA提取与PCR扩增
DNA提取:使用DNA富集beads进行全回收,80%乙醇清洗两遍,晾干2min左右,避免磁珠过于干燥导致回收率偏低,然后溶水回收
PCR扩增:使用包含测序平台(如Illumina)所需完整接头的引物进行PCR扩增。
04.文库构建
冰上配制扩增mix,主要为测序引物Index和扩增酶, PCR引物上带有独特的样本索引序列,使得多个样本的文库可以在同一个测序通道中进行混合测序,之后通过索引区分样本。
再次纯化:PCR后使用磁珠纯化扩增产物,去除引物二聚体等杂质。
使用高灵敏度仪器定量和质控文库,检测文库浓度和片段大小分布是否符合预期。
05.高通量测序
目的:对文库中的DNA片段进行大规模并行测序,获得片段两端的序列信息。
建议使用配对末端测序,可以获得更多的序列数据带来更好的比对结果,PCR重复的识别更加准确。
测序深度:取决于参考基因组的大小和预期的开放染色质程度
一般推荐:常规细胞系/组织样本:30-50 Million (M) 有效比对 reads
稀有细胞类型/单细胞ATAC数据整合:可能需要更深(>50M)
单细胞ATAC-seq:每个细胞通常几千到几万条reads,但总reads量巨大
四、金开瑞ATAC-seq实验数据分析
◆数据基本处理与质控:对原始数据进行过滤,去除接头信息、低质量碱基和未测出的碱基,获取有效数据(Clean Data)
◆文库插入片段长度分布
◆基因组测序深度累积分布
◆各样品基因promoter(up2K)区测序深度分布
◆Peak检测
◆Peak长度分布
◆Peak深度分布
◆样本生物学重复 IDR 分析
◆Peak在基因功能元件上的分布
◆Peak相关基因
◆Peak相关基因的GO功能显著性富集分析
◆Peak相关基因的pathway富集分析
◆样本间差异peak检测
◆样品间差异peak在基因功能元件的分布
◆样品间差异peak相关基因
◆样品间差异peak相关基因的GO和KEGG富集分析
五、案例分析
这篇文章发表在Nature Plants(IF:15.8),通过多组学策略(ATAC-seq、RNA-seq、CUT&Tag)揭示染色质动态与基因表达的协同作用。并确定了关键转录因子和潜在的转化效率增强子。
为了连接转录因子和靶基因,研究者使用 ATAC-seq 数据确定了足迹。大多数足迹分布在 ATAC-seq 峰的中间,宽度为 10 - 50 个碱基对,其中一半可以与拟南芥的转录因子结合基序相匹配。正如预期的那样,足迹的序列比基因间区域的序列更保守。对差异足迹的分析显示,参与植物再生的已知调节因子(如 LEC2、DOF5.6、WOX11 和 BBM)的转录因子活性发生了显著变化,表明它们潜在的功能重要性。
为了确定基因表达的时间顺序,研究者进行了拟时间分析,发现转录因子和靶基因在再生过程中表现出相似的转录谱。每个聚类中的转录因子调节同一聚类中的靶基因。在 C1(在第 0 天特别高表达)与其他聚类之间没有观察到调控关系,这进一步强调了生长素处理的影响。有趣的是,发现 C5 - C6 - C2 - C3 - C4 聚类之间的调控关系遵循基因表达的时间线。
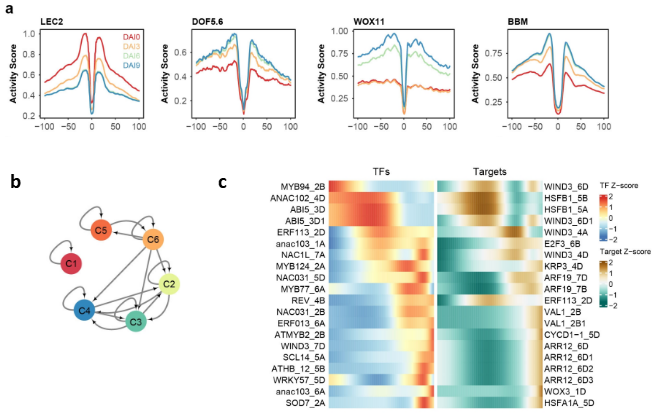
Figure a: 在不同感应阶段的LEC2,DOF5.6,WOX11和BBM的ATAC-SEQ足迹;活动评分反映了染色质的可及性。
Figure 4 b:不同的RNA簇之间的调节关系(Fisher的精确测试,p.adj < 1e-6))。
Figure 4c:TRN中TF和目标的表达模式, TFS与目标之间的调节关系已在拟南芥中证明。(来源于文献:Uncovering the transcriptional regulatory network involved in boosting wheat regeneration and transformation. Nat Plants)
ATAC-seq是一项革命性的技术,以其简便、灵敏、高分辨的特点,成为绘制染色质可及性图谱、解析基因调控网络的强大工具它在基础生物学、发育、疾病机制和精准医疗等领域发挥着越来越重要的作用。该技术无需抗体或复杂交联步骤,显著节省时间成本,而且可精准定位开放染色质区域(分辨率达单碱基水平),识别转录因子结合位点及核小体排布,适用于微量样本(低至500~50,000个细胞),尤其适合珍贵临床样本或单细胞分析。金开瑞可以提供ATAC-seq、ChIP-seq和CUT&Tag技术服务,期待与您携手合作,共同推动生命科学的发展。
最新动态
-
07.08
葡萄外泌体“激活细胞清道夫”——SM@G-ELNs协同水凝胶通过MERTK介导的巨噬细胞胞葬作用促进糖尿病伤口愈合(IF=14.3)
-
07.08
三篇顶刊论文,同一个技术选择——BiFC如何破解植物蛋白互作难题?
-
06.24
再登《Cancer Research》!金开瑞双荧光素酶报告系统助力机制研究新突破
-
06.24
登上《Gut》(IF 26.8)!南方医科大学团队揭示葛根类外泌体纳米囊泡靶向肠道细菌治疗类风湿关节炎新机制
-
06.18
亚细胞定位预测与验证结果不一致的原因是什么?
-
06.18
无需抗体、跨物种通用:三篇高水平论文实证DAP-seq如何助力植物转录调控?
-
06.11
打破“单向用药”的局限!IF=11.9《Asian J Pharm Sci》证实:红参外泌体,口服就能双向调控骨代谢
-
06.11
植物外泌体研究前沿:2026年4—5月6项突破性成果系统梳理
-
06.11
IF 26.8!颠覆“越小越好”的认知:南方医科大团队用40微米的柠檬胶囊,让肠屏障主动“开门”抗癌
-
06.11
从零开始研究一个基因,这篇讲透了!
 X
X 






