
分子动力学模拟(MD)结果怎么读?这份图表解读指南请收好!
一、技术背景概述
分子动力学(Molecular Dynamics, MD)模拟是研究蛋白质-配体复合物动态行为的强大工具,就像用计算机给原子和分子“拍动画片”——根据物理规则(比如原子间的吸引或排斥),计算每个原子每分每秒的位置和速度,把它们的运动过程连起来,就能看到微观世界里的“慢动作镜头”。通过MD模拟,我们可以了解:复合物是否稳定、关键相互作用是否持续存在、以及哪些氨基酸残基在结合中起核心作用。
二、应用方向
◆药物筛选与优化
研究药物与靶标蛋白的结合模式与亲和力,预测结合稳定性及耐药机制,指导药物开发。
◆蛋白质结构与功能研究
研究蛋白折叠、构象变化(如酶活性、信号传导、异常聚集等)及突变影响,揭示疾病机制与干预靶点。
◆核酸结构与功能研究
研究DNA/RNA构象变化、药物对核酸的干扰、核酸-蛋白互作(如转录因子、修复酶)、基因编辑机制及核酸药物可行性。
◆实验指导设计
基于分子动力学模拟,验证突变体设计及虚拟筛选分子的可行性。
三、核心结果图表解读
以下为分子动力学模拟中常见的分析图表及其解读方法。理解这些图表是判断模拟质量、提取生物学结论的关键。
1、均方根偏差(RMSD)曲线
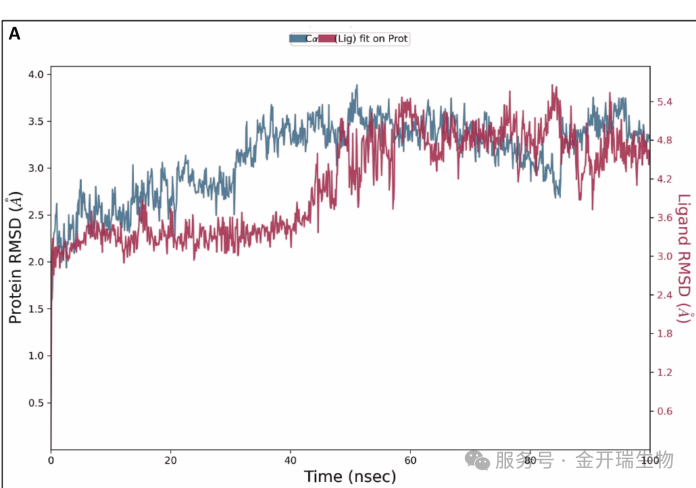
RMSD 的两条曲线
图表形式:横坐标为模拟时间(通常为纳秒),纵坐标为RMSD值(单位:Å或nm)。图中通常包含两条曲线:一条代表蛋白质主链(如Cα原子),另一条代表配体。
含义:RMSD衡量每一时刻的结构相对于初始参考结构的平均偏离程度。它是评估体系是否达到平衡稳定的首要指标。
如何看图?
|
RMSD 范围 |
稳定性评价 |
说明 |
|
< 2.0 Å |
★★★★★ 极度稳定 |
构象几乎无变化 |
|
2.0-3.0 Å |
★★★★ 稳定 |
正常波动,良好结合 → AP8ii (前 50ns) |
|
3.0-5.0 Å |
★★★ 可接受 |
有一定波动但整体稳定 → AP8ii (60-100ns) |
|
> 5.0 Å |
★★ 不稳定 |
构象显著变化或配体解离 |
平台期判断:当曲线在模拟后半段(如50 ns之后)围绕某一平均值小幅波动,不再持续上升,说明体系已收敛至稳定构象。
数值范围:对于蛋白质,RMSD稳定在2‑3 Å以内通常认为结构稳定;若超过5 Å,提示可能发生了较大的构象变化或部分去折叠。
配体曲线:配体RMSD应保持在3 Å以内,表示其在结合口袋中稳定结合。若配体RMSD持续上升并远离蛋白曲线,提示配体可能发生脱离或结合模式改变。
曲线对比:若蛋白与配体RMSD同时升高但保持同步,可能表示整体构象重排而非结合丧失。
示例解读:在100 ns模拟中,前60 ns蛋白RMSD逐步上升至2.5 Å,在 60 和 100 ns 之间的轨迹中 RMSD 突然增加,可能是由于初始蛋白-配体相互作用后的构象变化。值得注意的是,没有观察到实质性的结构改变,肽的构象在 CCR8 活性位点内保持一致取向。
2、均方根波动(RMSF)图
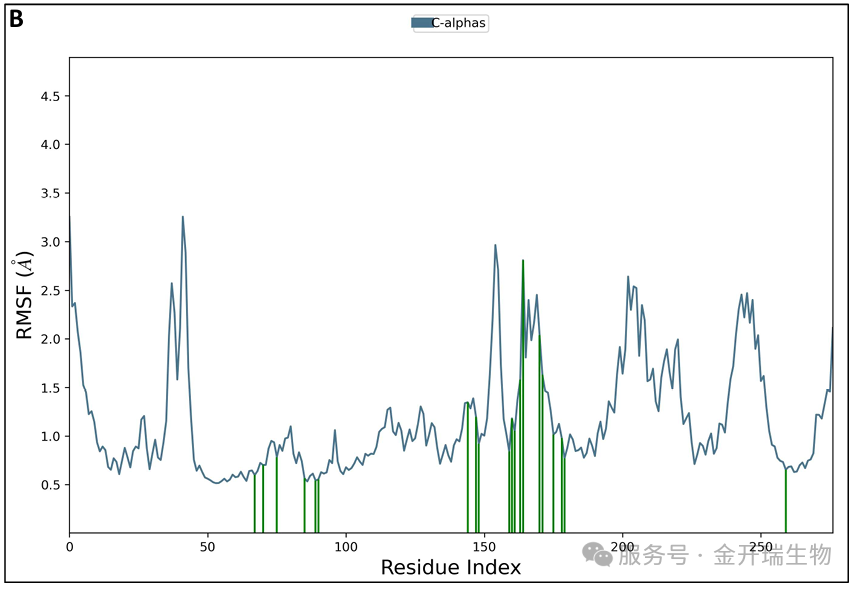
100 ns分子动力学模拟中CCR8蛋白的RMSF图
图表形式:横坐标为氨基酸残基序号(按序列顺序),纵坐标为每个残基的RMSF值(Å)。图中可能用绿色竖线标记与配体有接触的残基。
含义:RMSF (Root Mean Square Fluctuation, 根均方波动) 测量单个残基在模拟过程中的柔性/波动程度。与 RMSD 测量整体偏移不同,RMSF 关注每个氨基酸的“活跃度”。
如何看图?
识别柔性区域:峰值通常出现在loop区、末端(N端/C端),这是正常现象。α螺旋和β折叠区域的RMSF一般较低。
活性位点特征:与配体结合的关键残基通常表现为低RMSF(刚性),因为结合限制了其运动。若活性位点残基RMSF异常高,可能提示结合不稳定或模拟设置问题。
突变影响评估:比较野生型与突变体的RMSF图,若突变导致活性位点附近RMSF显著升高,可能削弱结合亲和力。
绿色竖条:图中标记的绿色竖条表示与配体有接触的残基。观察这些残基的RMSF值是否普遍较低,可验证结合界面的稳定性。
示例解读:
活性位点残基 (Glu286, Tyr113) 等:RMSF 值较低,表明在 AP8ii 结合后保持刚性
Loop 区域 RMSF 较高,这是正常现象,表明活性位点的低 RMSF 支持 AP8ii 稳定结合的结论。
3、回转半径(Rg)曲线
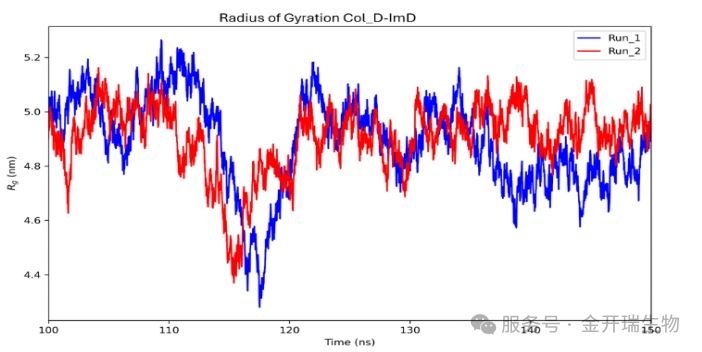
回转半径(Rg)曲线
图表形式:横坐标为模拟时间,纵坐标为Rg值(nm)。
含义:Rg衡量分子整体的紧凑程度,即所有原子质量中心到各原子的平均距离。Rg越小,结构越紧密;Rg越大,结构越松散或展开。它可以帮助我们理解分子在模拟过程中的折叠和展开行为。
如何看图?
趋势判断:若Rg在模拟过程中保持稳定,说明蛋白整体结构紧凑;若Rg逐渐增大,提示蛋白可能发生展开或构象松弛。
事件关联:当RMSD显示大幅波动时,若同时伴随Rg升高,可能表示蛋白发生了明显的构象重排或部分去折叠。
配体结合影响:配体结合后若Rg较未结合状态略有降低,提示结合可能诱导蛋白变得更加紧密。
4、溶剂可及表面积(SASA)曲线
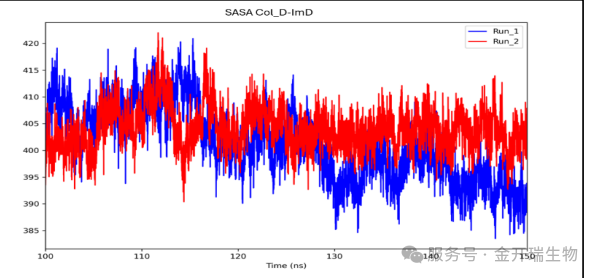
溶剂可及表面积(SASA)曲线
图表形式:横坐标为模拟时间,纵坐标为SASA值(nm²或Ų)。
含义:SASA表示分子表面可被溶剂分子(通常为水)接触的面积。它反映分子的疏水包埋程度及配体结合导致的表面变化。
如何看图?
结合事件:当配体结合到蛋白上时,配体占据部分表面,通常会导致蛋白SASA轻微下降(配体遮挡了原本暴露的区域)。
构象变化:若SASA在模拟中持续增加,提示蛋白可能正在展开,疏水核心暴露。
稳定性判断:稳定的SASA曲线(小幅波动)说明蛋白表面状态平衡。
5、自由能主成分分析图
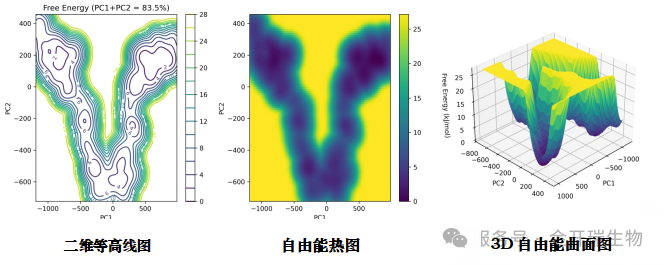
本组合图属于分子模拟与构象分析领域,是基于主成分分析(PCA)降维处理后得到的自由能可视化组合图。三张图采用不同可视化方式展示同一套二维降维后的自由能数据,所有子图统一以降维得到的第一主成分(PC1)为X轴,第二主成分(PC2)为Y轴,核心用途是分析目标分子体系不同构象的稳定性与能量分布特征。
标准解读流程:
第一步:交叉验证定位最稳定构象
需要结合三张图的特征交叉定位能量最低点:在等高线图中查找最中心的闭合等高线区域,在热图中查找颜色
最深的区域,在3D曲面图中查找谷底位置,三者重合的位置对应的构象就是体系的最稳定构象。
第二步:分析能量分布与构象状态
根据低能量区域的分布特征判断体系构象状态:
1.如果整个投影平面中只有一个连续的深色低能量区域,说明该体系仅存在单一稳定构象状态
2.如果投影平面中存在多个相互分离的深色低能量区域,说明体系存在多个可相互转换的稳定构象状态,不同构象之间存在可观测的能垒分隔。
第三步:验证结果可靠性
解读完成后需要核对左上角标注的累计贡献率:若数值<50%,说明当前降维结果无法很好代表体系真实结构变化,所得结论可能不可靠,需要调整PCA分析参数或改进降维方法重新计算。
6、自由能地貌图(FEL)状
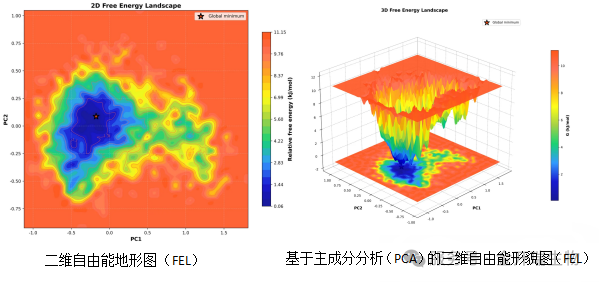
图表形式:
分析对象:基于主成分分析(PCA)的自由能地形图(FEL),包括二维等高线投影图与三维曲面图。
横坐标:第一主成分(PC1),反映模拟轨迹中最大的构象变化模式。
纵坐标:第二主成分(PC2),反映次大的构象变化模式。
颜色映射:从深蓝/紫色(低能量,稳定)到红/黄色(高能量,不稳定)。
标注信息:图中标有“Global minimum” (全局最小值),对应最低自由能区域。
含义:将模拟轨迹中所有构象投影到低维空间,计算每个区域的自由能,直观展示体系可能采用的稳定构象簇。
二维等高线图解读:
深蓝色区域(颜色条最低值约1.00)为全局最小值,对应最稳定构象。
等高线呈单一闭合椭圆,无其他分离的蓝色低谷→ 构象分布集中,无竞争性稳定态。
从深蓝向外颜色渐浅,能量梯度明显→ 稳定构象具有热力学优势,自发跳出盆地的概率低。
三维形貌图解读:
三维曲面中全局最小值处为一个明显凹陷的盆地,周围能量隆起(颜色转红/黄)。
盆地深度大、水平宽度窄→ 构象刚性较强,涨落幅度小。
无其他深度相近的盆地→ 体系在模拟时间内保持单一构象簇,未发生开闭运动或折叠/去折叠。
7、结合自由能分解(MM/PBSA残基分解)柱状图
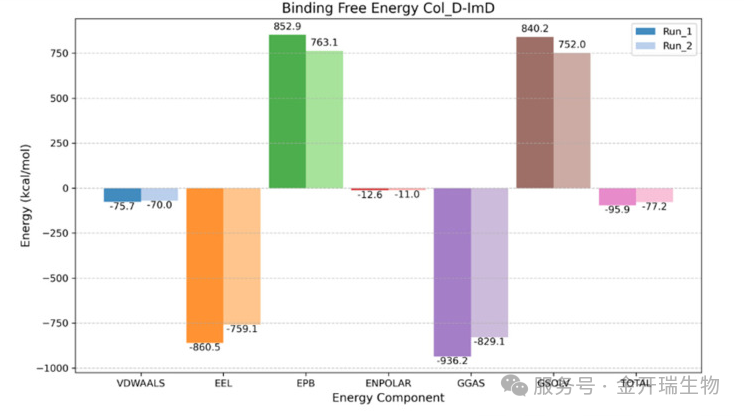
MM/PBSA 能量分解- 结合能柱状图
图表形式:
横坐标:能量组分,包括范德华能(VDWAALS)、静电相互作用能(EEL)、极性溶剂化能(EPB)、非极性溶剂化能(ENPOLAR)、气相总能量(GGAS)、溶剂化总能量(GSOLV)以及总结合自由能(TOTAL)。
纵坐标:能量值(单位:kcal/mol),正值表示不利贡献,负值表示有利贡献。
数据分组:两组独立模拟(Run_1 和 Run_2),每组用不同颜色柱状条并排显示。
含义:将总结合自由能分解到每个残基上,识别哪些氨基酸对结合起主要驱动作用。
如何看图?
该图展示了蛋白‑配体复合物结合自由能的总量及各能量项的分解。其中:
GGAS = VDWAALS + EEL,代表气相中分子间的相互作用(有利结合通常为负值)。
GSOLV = EPB + ENPOLAR,代表溶剂化效应(通常为正值,表示去溶剂化惩罚;但图中GSOLV为负值,说明溶剂化整体有利于结合,较为少见,需结合体系极性判断)。
TOTAL = GGAS + GSOLV,即总结合自由能,负值越大表示结合越强
该图表明两次独立模拟的总结合自由能分别为‑85.3和‑80.0 kcal/mol,结合主要依赖极强的静电相互作用(EEL约‑800 kcal/mol),范德华能起辅助作用,溶剂化效应整体仍促进结合;结果重复性良好,表明该复合物具有稳定且高亲和力的结合模式。
四、典型分析流程示例
以100 ns的蛋白‑配体复合物模拟为例,常规分析顺序及判断逻辑如下:
➤查看RMSD曲线:确认体系是否在合理时间内(如60 ns后)达到平台。若一直上升,需延长模拟时间或检查初始结构是否合理。
➤查看RMSF图:确认活性位点残基RMSF较低;若偏高,怀疑结合不稳定。
➤查看氢键分析:统计关键氢键的占据率,若低于30%,结合可能较弱。
➤计算MM/PBSA:获得总结合自由能及残基分解,识别热点残基,与RMSF
低值区域对比验证。
➤查看自由能地貌图:确认轨迹是否集中在低能区域,并提取代表性构象。
➤检查二级结构变化:确认关键功能区域未发生非预期结构转变。
五、模拟质量控制的常见考量
重复模拟:建议进行至少3次独立重复模拟(不同初始速度),以评估结果的统计方差。重复次数及分析深度可根据客户研究目标及期刊要求进行定制。
模拟时长:100 ns适用于多数柔性较小的体系;对于loop区丰富或构象变化大的体系,建议延长至250‑500 ns或更多。
力场选择:根据体系类型选择合适的力场(蛋白常用AMBER ff14SB、CHARMM36m;小分子用GAFF等)。
六、总结
分子动力学模拟提供的不仅是静态结构,更是一套描述分子动态行为的定量数据。正确解读RMSD、RMSF、Rg、氢键、自由能分解等图表,是判断模拟可靠性、提取生物学结论的基础。在实际应用中,应结合多个指标交叉验证,避免单一指标的误判。
金开瑞生物凭借专业的生物信息学分析平台和技术团队,可提供从分子对接、分子动力学模拟、结合自由能分析以及综合分析及课题整体打包服务,无论您是只需分子对接完成快速筛选,还是需要完整的“对接+MD+自由能”深度解析,我们都能灵活匹配您的需求,助力科研高效推进。
最新动态
-
07.08
葡萄外泌体“激活细胞清道夫”——SM@G-ELNs协同水凝胶通过MERTK介导的巨噬细胞胞葬作用促进糖尿病伤口愈合(IF=14.3)
-
07.08
三篇顶刊论文,同一个技术选择——BiFC如何破解植物蛋白互作难题?
-
06.24
再登《Cancer Research》!金开瑞双荧光素酶报告系统助力机制研究新突破
-
06.24
登上《Gut》(IF 26.8)!南方医科大学团队揭示葛根类外泌体纳米囊泡靶向肠道细菌治疗类风湿关节炎新机制
-
06.18
亚细胞定位预测与验证结果不一致的原因是什么?
-
06.18
无需抗体、跨物种通用:三篇高水平论文实证DAP-seq如何助力植物转录调控?
-
06.11
打破“单向用药”的局限!IF=11.9《Asian J Pharm Sci》证实:红参外泌体,口服就能双向调控骨代谢
-
06.11
植物外泌体研究前沿:2026年4—5月6项突破性成果系统梳理
-
06.11
IF 26.8!颠覆“越小越好”的认知:南方医科大团队用40微米的柠檬胶囊,让肠屏障主动“开门”抗癌
-
06.11
从零开始研究一个基因,这篇讲透了!
 X
X 






