
SWATH-MS的那些事
信息来源:金开瑞 作者:genecreate 发布时间:2018-11-27 15:01:22
SWATH-MS基本概念
SWATH-MS检测通常依赖于质谱的一级快速扫描 (通常至少10 Hz),高分辨(通常MS2的FWHM分辨率至少15000)以及高质量精度 (至少50ppm) ,其常见配置为第一级质量分析器为四级杆 (Q)而二级质量分析器为飞行时间(TOF)或Orbitrap(图3A)。近年来在四级杆设计上的技术进步使其能够有效的在整个近乎方形的母离子隔离窗口中传输离子(图4B)。这个重要的改进保证了获取到的信号的强度,并因此被检测肽段的定量值不再会因为位于隔离窗口的不同位置而变化了。
在SWATH-MS数据采集过程中,质谱反复记录了整个色谱梯度时间内一系列MS2扫描信息。而每个采集循环中MS1扫描信息可以只记录一次或多次 (图3A)。 其中关键的一点就是所有的MS2扫描是如何进行的与这些MS1并无关系。在每个MS2扫描时,都会隔离出一个相当宽的母离子窗口。该母离子隔离窗口的m/z会逐步递增,直到完整扫描预设的整个质量范围(图3A)。为了保证足够快速的记录MS1和MS2以进行色谱峰提取和肽段定量,需要对MS2扫描数量,每个扫描的累积时间,母离子隔离窗口大小,肽段扫描质量范围等参数均需要进行优化权衡。比如,最早提出SWATH-MS的Gillet用的采集方案是32个相同的25m/z的母离子隔离窗口,覆盖了400到1200m/z的质量范围,每个扫描的累积时间是100ms (图3B)。
紧接着而来的一个重要结论就是如此大的母离子隔离窗口将会同时引入大量肽段,并将它们同时碎裂(图3C和1D)。取决于样品复杂度,这些窗口里会同时引入数十个甚至数百个共碎裂的肽段(Rost等,2012),从而产生了高度混合而卷积的MS2谱图。如此高度多路的MS2谱图无法再用传统的基于数据库匹配的DDA谱图搜索软件直接进行分析。因此SWATH-MS数据需要更先进的数据处理策略。当然,DDA和靶向质谱通常也要设置0.5到3个m/z的隔离窗口,使得其碎片谱图一样会有来自于多个母离子的卷积碎片谱峰,但毕竟这样的谱峰复杂度还是远远低于DIA的(Michalski等,2011; Wang等,2014)。解决方法之一称之为“peptide-centric scoring” (Ting等,2015)。在这项策略中,采集到的数据集会和一套已知肽段各类参数的数据集进行比对搜索。最常用的方法就是预先采集一套谱图数据库。该策略也被称之“targeted data extraction”,也是在最早发表的SWATH-MS文献中提出 (Gillet等,2012) ,而至今为止也是分析DIA数据的主要手段。总的来说,在SWATH-MS实验中,所有落在隔离窗口范围内的离子化的多肽都将被同时碎裂并将被系统化的、无歧视的一并记录下来。SWATH-MS数据的定性和定量分析依赖于从高度多路的MS2谱图中提取,因此非常依赖于先进的数据处理策略,比如peptide-centric scoring。SWATH-MS是对追求蛋白质组全覆盖的DDA技术和追求高可重现性的靶向蛋白质组的一种补充。
SWATH-MS的优势
从上面总结的SWATH-MS的原理可以推导出其可能的性能优势和限制,我们在表1中进行了总结。SWATH-MS 和其他DIA技术的优势场景及其近期流行的原因均在引文中总结(Doerr,2015) ,接下来,我们也对其优势进行讨论。
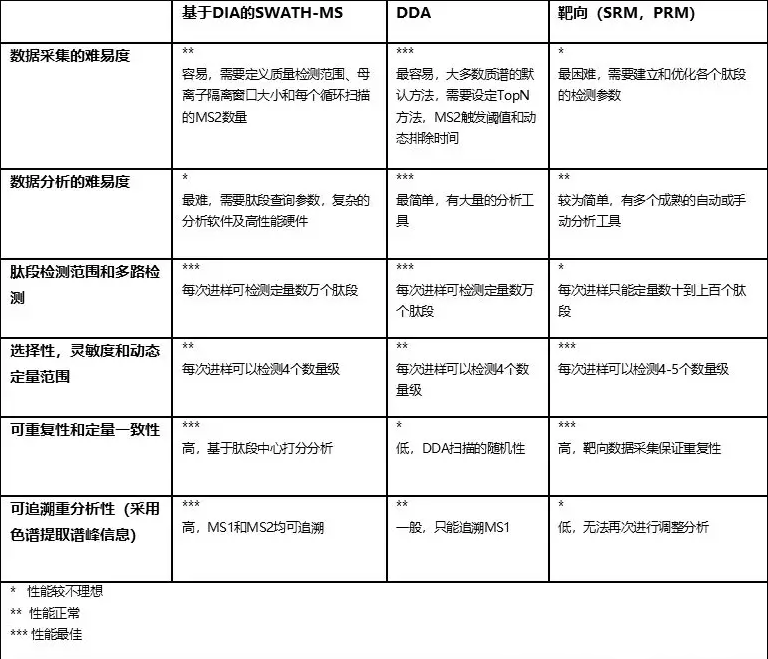
表1 优势和劣势:SWATH-MS和DDA及靶向(SRM,PRM)蛋白质组技术比较
1.数据采集的难易度
SWATH-MS的一项重要技术优势就是简化了数据采集流程。一次SWATH-MS采集需要定义母离子隔离方式(包括窗口峰宽、m/z采集范围,MS2扫描累积时间)。一旦方法确认,同一套系统中所有相似类型的样品均可采用统一的相同方法进行实验分析了。即使样品类型不同,同样的采集方法也被证明依然很可靠。这种数据采集方式的简便性和DDA采集差别不大,比靶向方法来得简单得多。像peptide precursor ions (PRM)或者peptide-fragment ion pairs (SRM)就需要预先定义每个感兴趣肽段的检测参数,且采集时经常需要确认好相应的保留时间 (Picotti & Aebersold,2012)。
2.肽段检测范围和多路检测
SWATH-MS的第二项优势是其对肽段定性和定量的高多路性能(一张MS2谱图含有多个肽段的碎片离子)。和基于DDA的方法类似,不同于靶向蛋白质组学,单次SWATH-MS检测可以定性超过10000种肽段。这早已在诸如人细胞系HEK293中进行了验证,通常这类样品在1%蛋白FDR水平下,2小时检测可以找30000到40000种多肽,对应于4000-5000种蛋白质 (Collins等,2017)。因此,相对于SRM或PRM方法,SWATH-MS实现了巨大的定量肽段数量提升。定量肽段越多,相应的可定量蛋白也越多,同时检测到的蛋白序列覆盖度也越高。这样可以改善蛋白组装的效果,相应带来更精确和准确的蛋白定量结果,进一步也可以帮助进行蛋白质翻译后修饰和可变剪切的检测。还有就是高多路检测率也可以提升全局数据归一化的效果。我们通常认为各个样品间大部分蛋白的浓度应该是不变的,但在SRM和PRM中,通常只检测目标肽段,因此按照这个逻辑进行样品间的全局归一化非常困难(尤其是目标肽段通常都是我们假定是参与的研究中生物学过程调控的蛋白)。SWATH-MS和DDA一样可以平行检测一系列在生物调控过程中无偏差的肽段,这是全局归一化的前提条件。
3.可重复性和定量一致性
当研究数百的样品的蛋白质组学时,肽段检测的可重复性及定量一致性就变成了主要困难。在DDA蛋白质组实验中要求肽段定量的高度一致性是非常困难的,因为你无法要求质谱DDA实时采集信号的时候所用的启发式母离子选择方式能够实现可重复性。因此当一个肽段在某次DDA检测中没有被定性成功,你不能判断该肽段到底是否可检出(“真阴性肽段定量”)。其实际情况很可能仅仅是因为质谱恰好因为一堆共流出肽段的存在,没有去碎裂这条多肽(“假阴性肽段定量”) (Michalski等,2011)。当然也有相应的降低这些假阴性的改进方法,比如尝试将某些样品中定性成功的肽段信息在不同样品中进行传递,从而大大改善定量数据矩阵的完整性 (Cox等,2014; Mueller等,2007; Prakash等,2006)。
相反的,在SRM,PRM和SWATH-MS中,MS2数据采集相当的有系统性且分析策略是以肽段为中心。通过这些方法的结合,我们可以较为可信的推导出目标肽段是的确“没被检测到”并因此推断出该样本中此肽段的含量至少是低于最低检测限的。尽管如此,当我们用自动化数据分析工具来检索数千种蛋白或数万种肽段时,SWATH-MS的 “假阴性肽段定量”在某种程度上依然存在。当然,和DDA一样,SWATH-MS的分析软件(Rost等,2016)也会进行样品间谱峰间对齐和鉴定结果转移。相对于母离子检测来说,碎片离子检测的灵敏度更高,且包含的信息也更丰富 (MS2中,肽段能够共流出大量碎片离子而MS1中一个肽段只能流出一个离子),因此这种结果转移方式相对于DDA来说更稳健、更灵敏。
最后一点,SRM (Abbatiello等,2015; Addona等,2009)和SWATH-MS (Collins等,2017)技术都被认为能够实现非常好的跨实验室和实验室内的结果重现性,且变异系数能够低于20% CV,即使在多个不同国家实验室进行的大规模研究也能达到这种程度。因此,如此高的重复性和一致性可以帮助生物学研究解决很多问题 (Rost等,2015)。
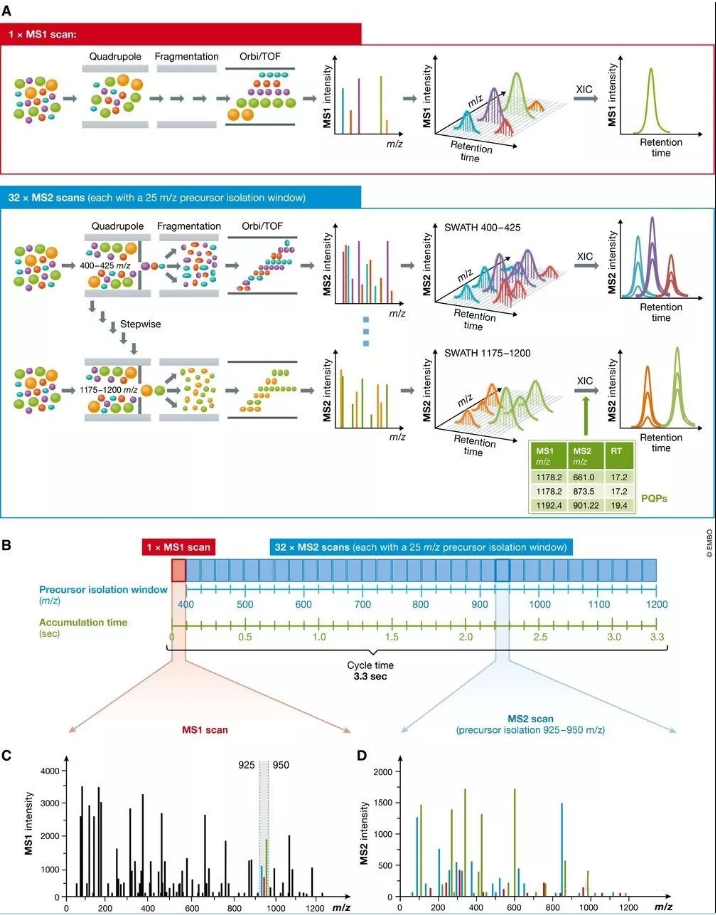
图3 SWATH-MS技术中顺序窗口数据非依赖采集的原理
(A) SWATH‐MS检测通常在具备快速质量扫描的杂交质谱上进行,主要在第一级质量分析器是四级杆而第二级质量分析器是TOF或Orbitrap的质谱上进行。在SWATH‐MS模式下,通常MS1扫描一次后会紧跟着一系列母离子隔离窗口很宽的MS2谱图(比如25 m/z的窗口大小)。通过在预先设定的质量范围内反复循环的连续母离子隔离扫描,质谱能够记录相对完整的谱图数据,其中包含了所有可检测的母离子和碎片离子的完整而连续谱峰信息。由此,MS1和MS2的提取离子色谱均可由此产生。而SWATH‐MS数据的分析则可以采用基于肽段的打分策略,需要预先记录所有可检索的肽段的色谱及质谱特征信息,从而合成一个由peptide query parameters (PQPs)构成的知识库来帮助进行肽段匹配。(B) 由Gillet et al (2012)提出的SWATH‐MS数据采集流程,在Q-TOF质谱上采集,采用预先设定的32个相同的25m/z的母离子隔离窗口,覆盖了400到1200m/z的质量范围,每个完整扫描的累积时间是100ms,完整的循环时间是3.3秒。 (C) 在某个时间点流出的MS1全扫描能够检测到的所有肽段母离子。比如质量范围在925到950 m/z,有三个共流出肽段被检测到(绿色,红色和蓝色标记)。 (D) 925–950 m/z范围内这3个肽段母离子产生的混合碎片离子MS2谱峰
4.可追溯查询
SWATH-MS数据的色谱和质谱的MS1母离子及MS2碎片离子谱峰信息都相当完整。这和DDA数据形成鲜明对比,DDA采集时只有母离子是连续记录而碎片信息则并不完整。SWATH-MS数据因此非常适合于过了一段时间后的追溯性分析,在最初的生物学假设中并不关注的信息或者早期无法确认的一些肽段分析参数,比如新获得的蛋白序列、肽段或者新的后修饰就可以再次进行检索分析(Rosenberger等,2017)。这不像SRM或PRM分析,如果要检测新的蛋白或肽段就必须得重新实验。
5.分析修饰肽段
这个优势是其对修饰肽段的定性和定量能力,它能够定位肽段序列中发生修饰的氨基酸位点,也能够搜索之前非预期的分析物。这些重要的能力是由SWATH-MS检测中对肽段整个流程时间内循环记录其高分辨、高精度MS2谱图所带来的收益。因此,不像DDA由于存在随机采集MS2和动态排除等问题,SWATH-MS可以将数据中的XIC相关MS1和MS2的信息最大化保留。还有就是对于PTM位点定位来说,碎片离子决定了修饰氨基酸的位置并且完整的MS2谱图可以用来判断不同可能位点间置信度差异(Keller等,2016; Meyer等,2017; Rosenberger等,2017)。最后一点,SWATH-MS和开放式修饰搜索工具结合可以发现非预期的全新修饰,且并不会造成搜索空间的组合爆炸 (Keller等,2016; Wang等,2015)。然后,当采用SWATH-MS来分析修饰肽段时也有和其他鸟枪法蛋白质组检测方式一样的固有挑战,亦即是修饰肽段大部分的浓度总是远远低于非修饰肽段。
总的来说,以上总结的SWATH-MS性能优势可以对复杂样本中海量的肽段进行精确定量,并能达到极高的重复性和一致性。典型的应用场景包括系统生物学研究 (Bensimon等,2012),基因相关的研究 (Liu等,2015; Okada等,2016; Williams等,2016) ,临床普筛(Liu等,2014; Sajic等,2015),药物/干扰组学普筛(Litichevskiy等,2017) 或者探索性的基础研究 (Collins等,2013; Lambert等,2013; Parker等,2015; Schubert等,2015b; Selevsek等,2015)。SWATH-MS同样适合进行快速检测分析,并能够单针检测就实现在复杂哺乳动物样本中约50%的蛋白质组覆盖率(Bruderer等,2017)。
SWATH-MS的缺陷和挑战
1.先验知识和预构建谱图库的获取难度
在最常规且成功应用SWATH-MS的分析流程中,肽段中心打分算法依赖于关于目标肽段的色谱、质谱特性先验知识所形成的高质量谱图库 (Schubert等,2015a)。通常谱图库由DDA方式产生,由此带来的问题就是只有DDA实验中能够检测到的肽段才可能在DIA中分析到。最近几年,不依赖于谱图库的SWATH-MS 数据分析替代方案也有了长足发展,比如DIA-Umpire (Tsou等,2015),FT-ARM (Weisbrod等,2012),和PECAN (Ting等,2017)。
2.数据分析的难度
目前,SWATH-MS 实验分析中主要的挑战就是肽段中心打分算法部分。高度多路的MS2谱图可能由多达10个到100多个共碎裂的肽段母离子形成,因此依赖于一套精巧的分析路线来检测和定量肽段并将它们进行可靠的统计分析。对于DDA蛋白质组学来说,20多年来已经发展了丰富的分析路线 (Cox & Mann,2008; Deutsch等,2015; Keller等,2005; Reinert & Kohlbacher,2010)。而靶向蛋白质组学分析软件也已经成熟应用了好几年了(Colangelo等,2013; Malmstrom等,2012; Teleman等,2012)。相应的自动化分析SWATH-MS肽段谱的软件还处于发展阶段,其中一些也已经得到了广泛应用并供免费使用(相应的综述参考 (Bilbao等,2015))。对其中五种广泛使用的软件的综合比较文献可以参考(Navarro等,2016)。
3.选择性,灵敏度和动态定量范围
SWATH-MS还有一个主要的挑战是其定量的态范围依然有限。多个课题组报道了在大规模复杂蛋白质组样本中碎片离子(MS2)具有比相应母离子(MS1)更高的信噪比,选择性和同一个扫描内的动态范围(Egertson等,2013; Gillet等,2012; Venable等,2004)。在复杂样本中,肽段的MS1信号很有可能被共流出的非常接近或相同m/z的信号所干扰。而在MS2水平的信号则仅来自于MS1隔离窗口内的这些母离子信号碎片,而由于MS2定量会选择多个离子碎片,其均受到干扰的可能性非常之低。另外考虑到离子阱质谱的选择性能力,MS1扫描的信号受到其AGC(auto gain control)效应的影响,高丰度肽段信号会抑制低丰度肽段,而SWATH-MS中由于只检测隔离窗口内的母离子碎片,该效应得到很好地改善。目前SWATH-MS在Q-TOF仪器上使用时,可以在单针检测复杂的注入人细胞系全酶解液的实验中覆盖4到4.5个数量级定量动态范围(Collins等,2017)。文献报道中的复杂样品定量最低检测限大约为中等attomole到低femtomole(上柱量)。如此定量范围还是比最新的SRM或MRM检测的灵敏度低3-10倍 (Gillet等,2012; Liu等,2013; Schmidlin等,2016) 。
综上,目前相对于SRM或PRM技术来说,SWATH-MS的主要劣势在于其灵敏度较低。因此,对于尤其关注低丰度肽段精确定量的项目来说,最佳的选择方案就选择一部分最感兴趣的目标肽段进行靶向定量。另外,大家也要注意SWATH-MS检测需要在谱图库和肽段查询流程建立和优化上花费较多的功夫。
最新动态
-
06.24
再登《Cancer Research》!金开瑞双荧光素酶报告系统助力机制研究新突破
-
06.24
登上《Gut》(IF 26.8)!南方医科大学团队揭示葛根类外泌体纳米囊泡靶向肠道细菌治疗类风湿关节炎新机制
-
06.18
亚细胞定位预测与验证结果不一致的原因是什么?
-
06.18
无需抗体、跨物种通用:三篇高水平论文实证DAP-seq如何助力植物转录调控?
-
06.11
打破“单向用药”的局限!IF=11.9《Asian J Pharm Sci》证实:红参外泌体,口服就能双向调控骨代谢
-
06.11
植物外泌体研究前沿:2026年4—5月6项突破性成果系统梳理
-
06.11
IF 26.8!颠覆“越小越好”的认知:南方医科大团队用40微米的柠檬胶囊,让肠屏障主动“开门”抗癌
-
06.11
从零开始研究一个基因,这篇讲透了!
-
06.11
解码RNA互作奥秘:金开瑞RIP/RNA pull-down试剂盒助力多篇高质量研究,深入解析肿瘤及椎间盘退变调控机制
-
06.11
别再大海捞针了!虚拟筛选正在改变药物发现的方式!
 X
X 






